Фенилкетонурия
Содержание:
Фенилкетонурия – диагностика
Фенилкетонурия у детей выявляется зачастую еще в роддоме, что позволяет вовремя начать лечение и не допустить развития ряда необратимых последствий. На 4-5 сутки после рождения у малышей берут капиллярную кровь натощак для определения некоторых тяжелых генетических заболеваний, среди которых — фенилкетонурия. Если выписка из роддома произошла раньше, анализ делают в поликлинике по месту жительства в течение первые 10 суток жизни.
С учетом того, что в редких случаях бывают ошибочные результаты, диагноз никогда не устанавливается по итогам первого анализа. Для подтверждения имеющейся патологии назначается ряд других исследований, среди которых:
- анализ мочи на выявление фенилпирувата;
- количественное определение фенилаланина и тирозина в плазме;
- определение активности печеночных ферментов;
- электроэнцефалография и магнитно-резонансная томография головного мозга.
Генетический дефект, приводящий к развитию патологии, может быть обнаружен у плода при инвазивной пренатальной диагностике. Для этого отбираются пробы клеток из ворсинок хориона либо амниотической жидкости, а затем проводится ДНК-анализ. Рекомендуется такое исследование в семьях с высоким риском заболеваемости, в том числе, если уже есть ребенок с фенилкетонурией.
Лечение фенилкетонурии
Главным способом лечения фенилкетонурии является диетотерапия, ограничивающая поступление в организм белка и фенилаланина.
Основным критерием адекватности диеты при фенилкетонурии служит уровень фенилаланина в крови, который должен:
- в раннем возрасте составлять 120–240 мкмоль/л;
- у детей дошкольного возраста – не превышать 360 мкмоль/л;
- у школьников – не превышать 480 мкмоль/л;
- у детей старшего школьного возраста допустимо увеличение содержания фенилаланина в крови до 600 мкмоль/л.
Пищевой рацион строится путем резкого ограничения поступления белковых продуктов животного и растительного происхождения и, следовательно, фенилаланина. Для облегчения расчетов принято считать, что 1 г условного белка содержит 50 мг фенилаланина.
При лечении фенилкетонурии полностью исключают продукты, богатые белком и фенилаланином: мясо, рыбу, сыр, творог, яйца, бобовые и др. В пищевой рацион больных входят овощи, фрукты, соки, а также специальные малобелковые продукты – амилофены.
Для коррекции белкового питания и восполнения недостатка аминокислот при фенилкетонурии назначаются специальные лечебные продукты:
- белковые гидролизаты: нофелан (Польша), апонти (США), лофенолак (США);
- смеси L-аминокислот, лишенные фенилаланина, но содержащие все другие незаменимые аминокислоты: фенил-фри (США), тетрафен (Россия), П-АМ универсальный (Великобритания).
Несмотря на обогащение аминокислотных смесей и белковых гидролизатов минеральными и другими веществами, больные фенилкетонурией нуждаются в дополнительном назначении витаминов, в частности группы В, минеральных соединений, особенно содержащих кальций и фосфор, препаратов железа и микроэлементов.
В последние годы для страдающих фенилкетонурией была обоснована необходимость применения препаратов карнитина (L-карнитин, элькар в средней суточной дозе 10–20 мг/кг массы в течение 1–2 мес. 3–4 курса в год) для профилактики его недостаточности.
Параллельно лечение фенилкетонурии осуществляется медикаментозным патогенетическим и симптоматическим лечением ноотропными средствами, препаратами, улучшающими сосудистую микроциркуляцию, по показаниям – антиконвульсантами.
Широко используется лечебная гимнастика, общий массаж и др. Комплексная реабилитация детей с фенилкетонурией предусматривает специальные методы педагогических воздействий в процессе подготовки к школе и школьного обучения. Больные нуждаются в помощи логопеда, педагога, в ряде случаев – дефектолога.
Большие споры вызывает вопрос о длительности диетотерапии в лечении фенилкетонурии. В последнее время большинство врачей принимает точку зрения о необходимости продолжительного выполнения диетических рекомендаций. Обследование детей, прекративших соблюдать диету в школьном возрасте, и детей, продолжавших получать диетотерапию, однозначно показало значительно более высокий уровень интеллектуального развития последних.
У больных фенилкетонурией старшего возраста, в том числе подростков, безусловно, возможно постепенное расширение диеты в связи с улучшением толерантности к фенилаланину. Коррекция питания осуществляется, как правило, путем введения в рацион ограниченного количества круп, молока и некоторых других натуральных продуктов, содержащих относительно умеренное количество фенилаланина. В период расширения рациона проводятся оценка нервно-психического статуса детей, контроль электроэнцефалограммы, уровня фенилаланина в крови.
В возрасте старше 18–20 лет проводится дальнейшее расширение диеты, однако и во взрослом периоде пациентам рекомендуется отказаться от высокобелковых продуктов животного происхождения.
Особенно строго подходят к диетотерапии девочек, страдающих фенилкетонурией, и женщин в репродуктивном периоде. Такого рода больным фенилкетонурией необходимо продолжать диетическое лечение для обеспечения рождения здорового потомства.
В последние годы разрабатывается способ снижения уровня фенилаланина в крови путем приема препарата, содержащего фенилаланингидроксилазу растительного происхождения.
Данная статья опирается на статью из книги «Врожденные и наследственные заболевания» под редакцией профессора П.В.Новикова, М., 2007
Ответы на часто задаваемые вопросы
Как проявляется фенилкетонурия у новорожденных?
Как выглядят больные фенилкетонурией?
- посветление волос и радужки глаза из-за недостатка пигмента меланина
- чрезмерная прибавка в весе
- быстро зарастает большой родничок
- суховатая кожа
- шелушение, сыпь и экзема
- частая рвота
- моча и пот с характерным «мышиным» запахом
- появляются судороги и спазмы
- скованность движений и зажатая «поза портного», что связанно с повышенным напряжением в мышцах
- неадекватное поведение, выкрики, смех
- уменьшение размеров черепа
- деформация ушных раковин
- дрожание пальцев рук
- недержание мочи
- выступающая вперед нижняя челюсть
Какие смеси использовать для ребенка с фенилкетонурией?
витаминымикроэлементыДля детей до одного года рекомендуют:
- Афенилак 13, Афенилак 15 от компании «Нутритек», Россия;
- MIDмил ФКУ 0 (Hero, Испания);
- ХР Аналог («Нутриция», Голландия);
- Фенил Фри 1 («Мид Джонсон» США).
Для детей старше одного года и для взрослых:
- П-АМ 1, П-АМ 2, П-АМ 3;
- Изифен (готовый продукт), а также ХР Максамейд и ХР Максамум с нейтральным и фруктовым вкусами («Нутриция», Голландия).
Какие бывают типы фенилкетонурии?
фенилкетонурии
- Фенилкетонурия I. Классическая и наиболее распространенная форма заболевания, описанная выше в статье. Связана с мутацией гена в 12-й хромосоме, при этом нарушается образование фермента фенилаланин-4-гидроксилазы, который превращает фенилаланин в тирозин.
- Фенилкетонурия II. При этой форме заболевания нарушение происходит в 4-й хромосоме. Нарушается выработка фермента дигидроптеридинредуктазы, который также способствует превращению фенилаланина в тирозин. Заболевание наследуется так же, как и I форма: для того, чтобы родился больной ребенок, необходимо, чтобы носителями гена были оба родителя. Распространенность фенилкетонурии II – 1 случай на 100 000 новорожденных.
- Фенилкетонурия III. В результате генетических нарушений возникает недостаток фермента 6-пирувоилтетрагидроптеринсинтазы. Наследуется, как и две предыдущие формы заболевания. Распространенность – 1 случай на 300 000 новорожденных.
Дают ли инвалидность при фенилкетонурии?
Критерии установления инвалидности при фенилкетонурии
- При фенилкетонурии I инвалидность устанавливают только при необратимых нарушениях со стороны центральной нервной системы, которые приводят к неврологическим расстройствам и умственной отсталости.
- При фенилкетонурии II и III типа группу инвалидности устанавливают во всех случаях.
Существует ли профилактика фенилкетонурии?
- Генетическое консультирование. Необходимо людям, планирующим завести ребенка, которые больны или являются носителями неправильного гена, у которых болен хотя бы один близкий родственник или уже родился больной ребенок. Консультирование проводит врач-генетик. Он помогает разобраться, как ген, ответственный за фенилкетонурию, передавался в предыдущих поколениях, каковы риски будущего ребенка. Также генетик помогает с планированием семьи.
- Скрининг новорожденных. Анализ не помогает предотвратить заболевание, но позволяет выявить его максимально рано, пока оно еще не привело к необратимым изменениям в головном мозге.
- Консультации и диета для женщин, страдающих фенилкетонурией. Если вы женщина и страдаете ФКУ, вам следует проконсультироваться с врачом и спросить, когда лучше планировать беременность в вашем случае. Во время беременности нужно соблюдать правильную диету – это помогает предотвратить дефекты развития у ребенка.
Каковы факторы риска фенилкетонурии?
- Как уже упоминалось в статье, ребенок рискует получить заболевание или стать носителем мутантного гена, если он есть у обоих родителей.
- Среди разных этнических групп распространенность фенилкетонурии различается. Например, среди представителей негроидной расы неправильный ген встречается реже.
- В группе повышенного риска находятся дети матерей, страдающих фенилкетонурией. Если во время беременности женщина не придерживается специальной диеты, у ребенка могут возникать дефекты развития.
Признаки и симптомы
Младенцы с фенилкетонурией обычно кажутся нормальными при рождении. При раннем обследовании и диетическом лечении у пораженных людей симптомы ФКУ могут никогда не проявляться. Однако нелеченные новорожденные с фенилкетонурией, которым не поставлен диагноз в первые дни жизни, могут быть слабыми и плохо питаться. Другие симптомы фенилкетонурии могут включать рвоту, раздражительность и/или красную кожную сыпь с небольшими прыщиками. Задержка развития может быть очевидна в возрасте нескольких месяцев. Средний IQ (коэффициент интеллекта) детей, не получающих лечения, обычно составляет менее 50. Умственная отсталость при ФКУ является прямым результатом повышенного уровня фенилаланина в головном мозге, который вызывает разрушение жирового покрытия (миелина) отдельных нервных волокон. Он также может вызывать депрессию из-за снижения уровня дофамина и серотонина (нейротрансмиттеров) в мозге.
У нелеченных младенцев с фенилкетонурией обычно необычно светлые глаза, кожа и волосы из-за высокого уровня фенилаланина, мешающего выработке меланина, вещества, вызывающего пигментацию. У них также может быть затхлый или «мускусный» запах тела, вызванный фенилуксусной кислотой в моче или потом.
У некоторых нелеченных пациентов с ФКУ присутствуют неврологические симптомы, включая:
- эпилепсию;
- мышечная ригидность;
- гиперрефлексию;
- тремор.
Не леченные женщины с фенилкетонурией, которые забеременели, имеют высокий риск выкидыша или проблем с ростом плода (задержка внутриутробного развития). У детей женщин с нелеченной фенилкетонурией могут быть аномально маленькая голова (микроцефалия), врожденные пороки сердца, аномалии развития или лицевые аномалии. Между тяжестью этих симптомов и высоким уровнем фенилаланина у матери существует тесная взаимосвязь. В результате все женщины с фенилкетонурией, прекратившие лечение, должны возобновить лечение до зачатия и продолжать его на протяжении всей беременности под контролем метаболического генетика и диетолога.
История
Фенилкетонурия как болезнь, протекающая с задержкой интеллектуального и психического развития, открыта в 1934 году норвежским ученым-исследователем Иваром Асбьером Феллингом, отсюда и другое обозначение патологии — болезнь Феллинга.
Первых успехов в коррекции здоровья малышей с ФКУ добились медики во главе с Хорстом Биккелем в середине прошлого века. Разработка терапии и ее внедрение проводились на базе Бирмингемского детского госпиталя (Англия).
Однако больших положительных результатов ученым удалось добиться, когда начала широко проводится ранняя диагностика новорожденных на выявление фенилкетонурии, это примерно 1958-1961 год прошлого века.
Расширение возможностей диагностики со временем позволило установить, что в развитии заболевания принимает участие только ген фенилаланингидроксилазы (РАН).
За последние десятилетия описаны атипичные варианты течения болезни, разработаны и широко применяются новейшие способы ее коррекции. В перспективе – использование генотерапии, что вполне вероятно позволит полностью победить болезнь.
Лечение фенилкетонурии
На сегодняшний день самым эффективным и распространенным способом лечения фенилкетонурии является элиминационная диета: диета с исключением продуктов, содержащих фенилаланин
Если ее строго придерживаться в первые годы жизни ребенка, когда развитие нервной системы еще продолжается, то можно вырастить здорового и полноценного человека. Очень важно исключение фенилаланина именно в первый год жизни, когда наиболее активно развивается нервная система. Если элиминационная диета назначается после года, умственные нарушения не излечиваются
Каждый месяц первого года жизни без применения диеты обходится ребенку безвозвратной потерей около 4 баллов IQ. Обычно достаточно придерживаться диеты до 16-18 лет, после этого возраста организм становится менее чувствительным к токсическому действию фенилаланина, и возможно расширение рациона питания. Включение новых продуктов необходимо проводить под контролем содержания фенилаланина в крови. Иногда требуется пожизненное строгое соблюдение диеты. Беременным женщинам и женщинам, планирующим беременность, и при этом больным фенилкетонурией, для рождения здорового ребенка обязательно строгое соблюдение диеты.
Степень строгости диеты зависит от концентрации фенилаланина в крови у ребенка. При его уровне до 2-6 мг% (120-360 мкмоль/л) диета не назначается, выше этого показателя – обязательна.
Суть диеты заключается в исключении белковых продуктов.
Отказ от грудного вскармливания не обязателен, но в этом случае кормящая мать должна строго придерживаться элиминационной диеты, потому что грудное молоко содержит белок (соответственно и фенилаланин). Вопрос о возможности грудного вскармливания решается индивидуально!!!
Для пополнения запасов белка назначают специальные смеси, не содержащие фенилаланин – Афенилак, Лофеналак, Нофемикс. После года это Фенилфри, Нофелан, Бигрофен, Тетрафен, МД мил ФКУ-3 и другие. В качестве прикорма назначают овощное и фруктовое пюре, фруктовые кисели, безбелковые каши (рисовая, кукурузная). После 6 месяцев можно применять специальные напитки Лопрофин, Нутриген и другие, кушать макаронные изделия, безбелковый хлеб.
В России обеспечение лечебным питанием детей, больных фенилкетонурией, по закону бесплатное.
Больным фенилкетонурией противопоказаны следующие продукты: мясо, рыба (и морепродукты), орехи, творог, твердый сыр, бобовые, яйца, изделия из пшеничной муки, гречневая и манная крупа, овсяные хлопья.
Во время назначения элиминационной диеты необходим строгий контроль содержания фенилаланина в крови: первые 3 месяца жизни – каждую неделю, от 3-х месяцев до года – минимум раз в месяц, от года до 3-х лет – 1 раз в 2 месяца. Стремятся к содержанию фенилаланина 2-6 мг% у младших детей, после 10 лет – до 10 мг%. Обязательно наблюдение у детского психоневролога.
Кроме элиминационной диеты периодически назначаются комплексы из витаминов и минералов. Если есть судорожные припадки, необходимо применение антиконвульсантов (Депакин, Клоназепам и другие). Многим из таких детей показан массаж, лечебная физкультура. Возможно использование средств физиотерапии для коррекции мышечного тонуса.
Атипичные формы фенилкетонурии не поддаются лечению элиминационной диетой. В этом случае показано применение гепатопротекторов, антиконвульсантов, препаратов с Леводопой (для коррекции гиперкинезов), 5-окситриптофана, Тетрагидробиоптерина (ВН 4). Эти формы фенилкетонурии имеют худший прогноз для жизни и тем более интеллектуального развития.
На сегодняшний день разрабатываются новые направления в лечении фенилкетонурии. Среди них стоит отметить следующие:
- использование заместительной терапии фенилаланинлиазой (PAL) – растительным ферментом, расщепляющим фенилаланин до нетоксических соединений;
- генная инженерия (введение искусственно созданного нормального гена, ответственного за фенилаланин-4-гидроксилазу);
- метод «больших нейтральных аминокислот» — уменьшение всасывания фенилаланина из пищи и поступления в головной мозг с помощью специальных препаратов.
Пока эти современные разработки не имеют широкого применения, но некоторые исследования, подтверждающие их эффективность, уже проводятся.
Как развивается фенилкетонурия
Врожденное наследственное заболевание, вызываемое нарушением обмена аминокислот, называется фенилкетонурией. Другие названия этой патологии – болезнь Феллинга (по имени впервые описавшего болезнь врача) или фенилпировиноградная олигофрения (основными последствиями прогрессирования заболевания являются тяжелые поражения центральной нервной системы и головного мозга, провоцирующие задержки развития, умственную отсталость).
В России фенилкетонурия регистрируется с частотой 1 случай на каждые 5-10 тыс. новорожденных, в Турции эта пропорция составляет 1:2500, в Японии и некоторых странах Европы – 1:100 000, в странах Африки заболевание практически не встречается. По статистике девочки наследуют патологию в два раза чаще, чем мальчики. В неонатальном периоде клинические проявления болезни отсутствуют, но из-за поступления фенилаланина в организм с пищей манифестация фенилкетонурии происходит в первые полгода жизни.
При болезни Феллинга дефицит печеночного фермента фенилаланин-4-гидроксилазы вызывает нарушения метаболизма фенилаланина, поступающего с белковой пищей. Происходят следующие процессы:
- Запускаются побочные пути окисления фенилаланина, при которых в организме накапливаются его токсичные производные – фенилуксусная, фенилмолочная и фенилпировиноградная кислоты.
- Возникает дефицит тирозина (аминокислота, продукт нормального метаболизма фенилаланина), участвующего в функциях гипофиза, надпочечниках и щитовидной железы, выработке меланина, регулирующего аппетит и процессы жировых отложений.
- Развиваются нарушения обмена липопротеидов, гликопротеидов, белков.
- Происходит расстройство транспорта аминокислот.
- Нарушается обмен катехоламинов и серотонина.
- Образуется избыток фениэтиламина и ортофенилацетата, провоцирующий нарушения липидного метаболизма в головном мозге.
- Разрушение механизма передачи импульсов между клетками нервной системы из-за возникающего дефицита нейромедиаторов.
Причины
Синдром фенилкетонурия у детей в 98% случаев развивается вследствие мутации расположенного на длинном плече 12 хромосомы гена, кодирующего количество фенилаланингидроксилазы (что приводит к недостаточности этого фермента). Генетическая патология носит аутосомно-рецессивный характер наследования, то есть для развития болезни новорожденный должен унаследовать копию дефектного мутировавшего гена и от отца, и от матери, являющихся его гетерозиготным носителем. Этим обусловлена низкая степень распространенности патологии.

Формы и типы
Классическая фенилкетонурия 1 типа, развивающаяся в результате наследования от обоих родителей мутировавшего гена, составляет 98-99% от всех зарегистрированных случаев. Другими, атипичными формами болезни, не поддающимися лечению диетотерапией, но протекающими с аналогичными клиническими проявлениями, являются:
- фенилкетонурия 2 типа, развивающаяся в результате дефицита дегидроптеринредуктазы;
- фенилкетонурия 3 типа, возникающая из-за недостаточности тетрагидробиоптерина.
Симптомы
 У детей с фенилкетонурией светлые волосы, бледная кожа и голубые глаза.
У детей с фенилкетонурией светлые волосы, бледная кожа и голубые глаза.
Ребенок с фенилкетонурией рождается внешне здоровым, то есть ничем не отличается от других детей. С поступлением пищи в организм начинается попадание белка, а значит и фенилаланина. Последний постепенно накапливается, и обычно к 2 месяцам жизни появляются первые симптомы: вялость или беспокойство, отсутствие интереса к окружающему миру, срыгивания, изменения мышечного тонуса. Иногда срыгивания столь частые и обильные, что возникает подозрение на патологию желудочно-кишечного тракта (пилоростеноз). Ребенок из-за срыгиваний может плохо набирать в весе.
К 4-6 месяцам становится очевидной задержка психического развития. Ребенок не следит за игрушкой, не реагирует на звук, не узнает родителей. Чем дольше продолжается поступление фенилаланина в организм с едой, тем выраженнее нарушения в психической и мыслительной сферах. Развитие речи резко задерживается. Иногда словарный запас может ограничиваться несколькими словами. Если диагноз не будет выставлен и не будет начато лечение, то к 3-4 годам умственные нарушения достигнут степени идиотии (самая тяжелая степень олигофрении).
Особенностью клинического течения фенилкетонурии является необратимость возникших психических и интеллектуальных изменений. То есть при позднем выявлении помочь таким деткам уже нельзя – на всю жизнь они остаются умственно отсталыми.
Физическое развитие также отстает: дети позже начинают держать голову, переворачиваться, сидеть. Когда такие дети начинают ходить, то при этом они широко расставляют ножки, сгибая их одновременно в коленных и тазобедренных суставах. Походка покачивающаяся, мелкими шажками. В положении сидя дети принимают «позу портного» — сгибают и руки, и ноги, поджимая последние под себя. Обычно объем головы меньше, чем в норме. Может быть выраженная микроцефалия: маленькая голова.
Из других неврологических симптомов возможны нарушения мышечного тонуса, судорожные припадки. Эпилептические приступы обычно появляются в возрасте 1,5 лет и приводят к еще большему прогрессированию нарушений интеллекта.
У части больных фенилкетонурией появляются непроизвольные движения в конечностях, дрожание (гиперкинезы). В движениях нет плавности и согласованности, нарушается равновесие.
Кроме ряда психических и интеллектуальных изменений, фенилкетонурию характеризуют следующие симптомы:
- специфический «мышиный» запах (или запах плесени) от ребенка: этот симптом характерен только для фенилкетонурии. Запах появляется в результате выделения продуктов метаболизма фенилаланина (фенилпировиноградной, фенилмолочной, фенилуксусной кислот) через кожу и с мочой;
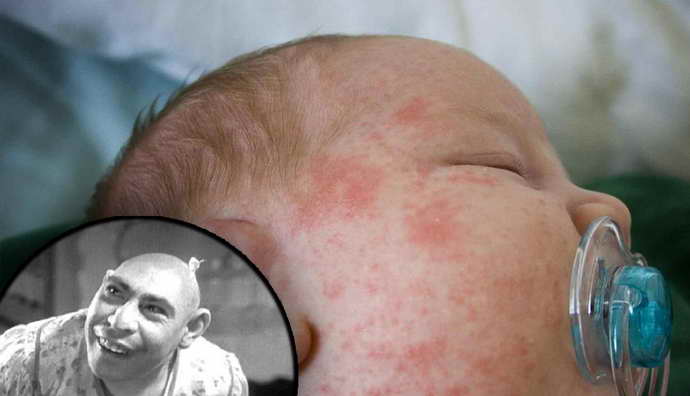
- кожные проявления: дерматиты, экзема, просто шелушение (возникают по той же причине, что и «мышиный» запах);
- позднее прорезывание зубов: у таких детей первые зубы могут появиться после 18 месяцев, эмаль недоразвита;
- нарушение пигментации: у таких детей обычно голубые глаза, очень светлая кожа и волосы в результате снижения количества меланина (его содержание зависит от метаболизма фенилаланина). Из-за этого у таких детей наблюдается повышенная чувствительность к солнечному свету;
- вегетативные симптомы: пониженное артериальное давление, повышенная потливость, запоры, акроцианоз (синюшность кистей и стоп);
- нередко фенилкетонурия сопровождается врожденными пороками сердца.
Атипичные случаи фенилкетонурии, связанные с нарушением деятельности других ферментов, участвующих в метаболизме фенилаланина, кроме умственных изменений характеризуются развитием мышечной слабости во всех конечностях с одновременным повышением мышечного тонуса, спастическим тетрапарезом. Также при этих формах развивается слюнотечение, приступы повышения температуры.
У взрослых людей, страдающих фенилкетонурией, возможно появление судорожных припадков, нарушений координации, дрожания в конечностях, ухудшения памяти и внимания, возникновение депрессии. Обычно подобные симптомы возникают при несоблюдении элиминационной диеты.
Фенилкетонурия – что это за болезнь?
Фенилкетонурия, или болезнь Феллинга, является тяжелой патологией, впервые описанной в 1934 году норвежским ученым Феллингом. Тогда Феллинг провел обследование нескольких детей с умственной отсталостью и выявил у них присутствие в моче фенилпирувата – продукта распада поступающей с едой аминокислоты фениланина, которая в организме больных не расщепляется. Фенилкетонурия – заболевание, связанное с нарушением обмена веществ врожденного характера, открытое одним из первых.
Фенилкетонурия – тип наследования
Болезнь Феллинга является хромосомно-генетической, наследственной, передаваемой детям от родителей. Виновником развития патологии выступает ген, находящийся на 12 хромосоме. Он ответственен за производство печеночного фермента фенилаланин-4-гидроксилазы, за счет которого происходит превращение фенилаланина в другое вещество – тирозин (оно требуется для нормальной работы организма).
Установлено, что фенилкетонурия наследуется как рецессивный признак. Приблизительно 2 % людей являются носителями дефектного гена, но при этом не страдают фенилкетонурией. Патология развивает только тогда, когда и мать, и отец передают ген ребенку, а случиться это может с вероятностью 25 %. Если фенилкетонурия наследуется как рецессивный признак, жена гетерозиготна, а муж гомозиготен по нормальному аллелю гена, то вероятность того, что дети будут здоровыми носителями гена фенилкетонурии, равна 50 %.
Формы фенилкетонурии
Рассматривая, у кого может развиться фенилкетонурия, что это за заболевание, зачастую речь ведется о классической форме патологии, которая встречается примерно в 98 % случаев. Остальные случаи – кофакторная фенилкетонурия, обусловленная дефектом тетрагидробиоптерина вследствие нарушения его синтеза или восстановления активной формы. Данное вещество служит кофактором ряда ферментов, и без него невозможно проявление их активности.

Фенилкетонурия – причины
Болезнь Феллинга является патологией, при которой из-за мутаций в гене, вызывающих дефицит или отсутствие фенилаланин-4-гидроксилазы, происходит накопление в тканях и физиологических жидкостях фенилаланина, а также продуктов его неполного расщепления. Часть избыточного фенилаланина превращается в фенилкетоны, выводимые с мочой, что и обусловило название болезни.
Нарушение обменных процессов сказывается в большей степени на головном мозге. На его ткани производится отравляющее воздействие, нарушаются процессы жирового обмена, происходит сбой миелинизации нервных волокон, снижение образования нейромедиаторов. Так начинается запуск патогенетических механизмов задержки умственного развития у ребенка.
Симптомы классической формы фенилкетонурии
Общие проявления
Если неонатальный скрининг не был проведён в родильном доме, заболевание осталось не распознанным и не были организованы необходимые лечебные мероприятия, то ФКУ проявит себя на первом году жизни крохи. Первые признаки отклонений от нормы родители обнаружат у ребёнка в возрасте 2 – 6 месяцев, когда ранее здоровый карапуз становится вялым и апатичным или наоборот, чрезмерно раздражительным. Приём пищи часто заканчивается срыгиванием, а на коже младенца появляются проявления аллергического дерматита.
Характерная внешность
Поскольку нарушение обмена аминокислот приводит к уменьшению образования меланина, в случае развития заболевания кожные покровы, волосы, радужная оболочка глаз теряют пигмент. Малыши с генетическим заболеванием имеют светлые волосы, тонкую бледную кожу, голубые глаза. Указывать на наследственный недуг может характерный запах пота и мочи крохи. Многие врачи характеризуют его как «мышиный», а связан он с выделением продуктов обмена аминокислот с биологическими жидкостями.

Поражение нервной системы
Первые признаки изменений со стороны нервной системы проявляются вялостью, снижением тонуса мышц крохи. Нередко возникает дистония, появляются непроизвольные сокращения мышц, навязчивые движения, судороги. Возможно развитие органических патологий головного мозга – микроцефалии и гидроцефалии.
Практически у 50% больных фенилкетонурией развивается эпилепсия. Иногда характерные приступы являются первым признаком наследственного заболевания. Пароксизмы плохо поддаются лечению обычными противосудорожными препаратами и значительно снижают качество жизни ребёнка.
Отставание в развитии
С течением болезни малыш быстро теряет интерес к окружающему миру, становится апатичным. Физическое развитие ребёнка так же страдает – кроха поздно овладевает необходимыми навыками. Задержка в психическом развитии становится очевидной, когда возникают проблемы в формировании речи, отставание в умственном развитии.

Исследования показали, что грубые изменения интеллектуальной деятельности развиваются уже через год прогрессирования болезни. Кроха необратимо теряет до 50 баллов IQ ежегодно, что впоследствии приводит к развитию дебильности, имбецилии или идиотии. Поэтому сроки начала лечения фенилкетонурии очень важны.
Неврологические проблемы у детей старшего возраста выражаются в гиперактивности, появлении стереотипий, навязчивых движений кистей рук, лица, языка, покачиваний. Иногда расстройства психики проявляются в шизофреноподобных состояниях.
Грубое отставание в развитии приводит к отсутствию навыка ходьбы у 30% больных детей и алалии, недоразвитии речи, у 60% пациентов.
Причины

При классической форме заболевания, так называемой фенилкетонурии I типа, встречающейся в 98% всех диагностированных случаев, в основе лежит недостаток печеночного фермента, который поступает с пищей и участвует в превращении фенилаланина в тирозин (входит в состав пигмента, ферментов, необходим для функционирования всего организма). В результате концентрация фенилаланина в крови, спинномозговой жидкости увеличивается, соответственно, концентрация тирозина падает. Процессу сопутствует нарушение миелинизации нервных волокон, что ведет к уменьшению образования нейромедиаторов, которые участвуют в передаче нервных импульсов между клетками нервной системы, в результате запускается механизм отставания умственного развития.
Помимо классического типа, существуют атипичные формы болезни, причины фенилкетонурии в этом случае связаны с мутацией в других генах, которые ответственны за кодирование фермента. Вероятность рождения ребенка с подобной патологией у родителей-носителей мутантного гена составляет 1:4. Клинические проявления подобны симптомам при классической форме болезни, однако данная патология не поддается коррекции с помощью диеты.
Следует заметить, что риск развития патологии увеличивается при рождении ребенка у близких родственников. Для появления симптоматики ребенок должен получить от обоих родителей (носителей мутантного гена) по одной дефектной копии гена; такая вероятность в случае брака между ближайшими родственниками существенно возрастает. В этом случае заболеванию сопутствует глубокая умственная отсталость.
Симптомы заболевания
При своевременном обнаружении болезнь Феллинга поддается успешному лечению путем корректировки питания, и развитие ребенка происходит в соответствии его возрастной группе. Трудность выявления генной мутации заключается в том, что ранние признаки тяжело обнаружить даже опытному педиатру. Выраженность симптоматики врожденного заболевания усиливается по мере взросления ребенка, потому что употребление белковой пищи способствует развитию нарушений ЦНС.
Признаки у новорожденных
На протяжении первых дней жизни ребенка признаки патологических отклонений обнаружить трудно – малыш ведет себя естественно, задержки в развитии не наблюдается. Симптомы заболевания впервые начинают проявляться через 2-6 месяцев после рождения. Родителей должно насторожить поведение малыша, которое характеризуется низкой активностью, вялостью, или, наоборот, беспокойством, гипервозбудимостью.
С началом грудного вскармливания в организм новорожденного с молоком начинают поступать белки, что служит катализатором появления первых признаков, однозначно свидетельствующих о том, что заболевание начало прогрессировать. К специфическим клиническим проявлениям болезни относятся:
- постоянная рвота (зачастую принимаемая за врожденное сужение привратника);
- частое срыгивание;
- отсутствие реакции на внешние раздражители;
- мышечная дистония (сниженное напряжение мышц);
- судорожный синдром (судороги эпилептического или неэпилептического характера).
Симптомы у детей после 6 месяцев
Если манифестация генетической болезни не произошла (или не была замечена) в течение первых 6 месяцев с момента рождения ребенка, то после этого периода уже можно точно определить отставание в психомоторном развитии. Симптомами генетических нарушений, вызванных ферментным дефицитом, у детей, старше полугода, являются:
- снижение активности (вплоть до полной безучастности);
- отсутствие попыток к самостоятельному вставанию, сидению;
- особенный «мышиный» запах кожи (запах плесени возникает вследствие выведения токсических производных фенилаланина через потовые железы и мочу);
- потеря способности к визуальному распознаванию лиц родителей;
- шелушение кожи;
- появление дерматитов, экзем, склеродермии.
Прогрессирование заболевания при отсутствии лечения в детском возрасте
Если отклонения в развитии не были выявлены в младенческом возрасте, и соответствующее лечение не проводилось, то заболевание начинает активно прогрессировать и нередко приводит к инвалидности. Отсутствие терапии на раннем этапе болезни вызывает появление следующих симптомов болезни уже в возрасте 1,5 лет:
- микроцефалия (уменьшенные размеры головного мозга);
- прогнатия (смещение верхнего зубного ряда вперед);
- позднее прорезывание зубов;
- гипоплазия эмали (истончение или полное отсутствие зубной эмали);
- задержка речевого развития вплоть до полного отсутствия речи;
- 3, 4 степень олигофрении (задержка психического развития, умственная отсталость);
- врожденные пороки сердца (дефекты в структуре сердечной мышцы, отделах сердца, крупных сосудах);
- расстройства вегетативной системы (акроцианоз, повышенная потливость, артериальная гипотония);
- запоры.