Синдром эдвардса: клинические проявления и возможности терапии
Содержание:
Развитие детей: особенности синдрома Эдвардса
Только внешним видом при появлении на свет и проявлениями пороков в развития, аномалия Эдвардса не ограничивается, по мере роста и развития возможны проявления все новых и более тяжелых отклонений. Первые из них можно выявить через несколько недель с момента рождения. Нередко по ним впервые можно заподозрить диагноз при мозаичном и неполном типе патологии, если внешних дефектов мало или практически нет. Описанные выше пороки и внешние изменения становятся по мере роста более резкими и выраженными, постепенно добавляются новые проблемы и аномалии, которые при рождении не были заметны. Так, сюда стоит отнести:
- Резкое и выраженное отставание в уровне физического развития. При рождении масса таких детей не превышает 2-2.5 кг, и причиной тому являются хромосомные дефекты, которые не дают полноценно развиваться системам и органам. По мере развития показатели роста и массы тела по месяцам существенно отличаются от норм, также слабо прибывают и окружности груди. Размеры головы могут прибывать но норме или выше нее, что может указывать на скопление внутри полости черепа избытка жидкости, формируя гидроцефалию.
- Проблемы со стопами. По мере роста формируется косолапость в силу аномалий строения костей и связок, мышечного гипертонуса. Страдает и контроль за развитием стопы и тонусом от нервной системы. Это угрожает тем, что дети поздно начинают ходить или не могут этого делать вообще в силу общего тяжелого состояния. Внешне косолапость проявляется в выраженной деформации стопы и ненормальной ее установке в покое.
- Нарушение мышечного тонуса. При рождении гипертонус типичен для кистей, формируя их флексорное положение, но по мере развития распространяется на соседние групп мышц, приводя к вычурным позам. Но чаще тонус мышц снижен, дети вялые и атоничные, конечности весят плетьми. Могут также быть состояния дистонии с резким сокращением отдельных мышечных групп на фоне гипотонии других. При этом руки могут быть согнуты, а ноги – переразгибаются. Это приводит к нарушению координации и хаотичности движений, вывихам и ненормальному положению конечностей.
- Атипичные реакции нервной системы, проблемы эмоциональности. Некоторые отделы мозга у детей с синдромом Эдвардса недоразвиты, в связи с чем формируются проблемы эмоциональности и заторможенности, неадекватные реакции на окружающее. Это связано обычно с мозолистым телом и мозжечковыми гипоплазиями, что формирует также отставание умственного развития. Внешне это проявляется в отсутствующем взоре, неэмоциональности и невозможности установления контакта, отсутствии слежения за объектами. Дети не реагируют на звуки в силу проблем с ушами и нервной системой, подобные аномалии выявляют в первые же месяцы жизни.
В целом, в силу тяжелых и комбинированных поражений дети не доживают до периода совершеннолетия. Часть их при мозаичной форме может достигать возраста подростков с глубокими степенями имбецильности и внешними проявлениями пороков.
Проявления синдрома
Дети с трисомией 18 рождаются с низким весом, в среднем около 2200 грамм, при этом длительность беременности — нормальная или даже превышает норму. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и лицевого черепа, мозговой череп имеет долихоцефалическую форму. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Наружный слуховой проход сужен, иногда отсутствует. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80 % случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка), большой палец утолщён и укорочен. Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных сосудов: дефект межжелудочковой перегородки, аплазии одной створки клапанов аорты и лёгочной артерии. У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса, переходящее в повышение со спастикой.
Лечение синдрома Эдвардса
В настоящее время лекарств от синдрома Эдвардса не существует. Детки с таким диагнозом страдают от умственной и физической недоразвитости, и вопрос терапии буквально заводит врачей в тупик.
Некоторые проблемы и пороки помогает устранить хирургическое вмешательство, но инвазивное лечение неоправданно при детском возрасте, который едва достигает нескольких дней или месяцев. По этой причине специалисты склоняются к паллиативной терапевтической тактике. Она подразумевает поддержание – прежде всего, психологическое – деток, рожденных с подобными отклонениями. 5 – 10% малышей с синдромом Эдвардса доживают до годовалого возраста.
Нарушения деятельности нервной системы и мышечного аппарата приводят к расстройству двигательной активности. Повышается риск развития сколиоза, косоглазия, мышечной атрофии. Хирургическое лечение при данной болезни может быть ограничено по причине риска развития сердечнососудистых осложнений.
Такие детки страдают от частых запоров, являющихся следствием гипертонуса брюшной стенки и атонией кишечника. Вследствие этого грудные дети испытывают дискомфорт, а также имеют проблемы с приемом пищи. Для уменьшения выраженности симптоматики необходимо применять специальные молочные смеси, слабительные препараты и средства из разряда так называемых «пеногасителей». Категорически недопустимо прибегать к клизмированию, поскольку оно может нарушить электролитный баланс организма.
Для коррекции физического и умственного развития детей с данным синдромом разрабатываются специальные программы.
Неблагоприятно сказывается на прогнозе для ребенка образование опухоли «Вильмса», являющейся одним из видов рака почки. В связи с этим рекомендуется регулярное проведение УЗИ органов брюшной полости.
Больные дети часто подвергаются инфекционным заболеваниям и прочим патологическим состояниям наподобие:
- инфекционных поражений мочеполовой системы;
- среднего отита;
- конъюнктивитов;
- синуситов;
- фронтитов;
- пневмонии;
- ночного апноэ;
- легочной гипертензии;
- врожденных сердечных пороков;
- артериальной гипертензии.
Родители должны быть готовыми к такому повороту событий, чтобы своевременно отреагировать на развитие той или иной болезни, и незамедлительно обратиться за помощью к врачу. Параллельно с этим нужно тщательно наблюдать за общим состоянием больного ребенка, так как вовремя выявленное и подвергнутое терапии заболевание является залогом продления детской жизни.
Причины
Кариотип здорового ребенка состоит из 46 хромосом: по 23 от обоих родителей. У лиц с синдромом Эдвардса под влиянием не установленных наукой факторов происходит дублирование генетического материала, и появляется дополнительная 47 хромосома, которая является «лишней». Обычно мутации подвергается 18 хромосома. Так формируется название – трисомия 18. Хромосомные нарушения происходят в процессе образования гамет или дробления зиготы. В большинстве случаев возникает простая трисомия 18, крайне редко – мозаичная или транслокационная форма. Причины синдрома Эдвардса в настоящее время остаются неизвестными. Больной ребенок может появиться на свет в семье, где родители и родственники являются абсолютно здоровыми.
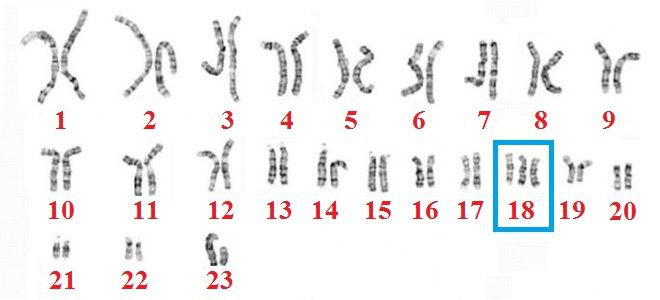
- Полная трисомия – три 18 хромосомы в каждой клетке плода. Патология обусловлена нерасхождением хромосом в процессе мейоза. Почти всегда лишняя хромосома передается по материнской линии. Этот вариант синдрома Эдвардса протекает довольно тяжело по сравнению с другими формами, встречается намного чаще и практически всегда имеет неблагоприятный прогноз.
- Мозаицизм связан с нерасхождением хромосом после слияния половых клеток на ранней стадии дробления зиготы. Обе гаметы изначально имеют нормальный набор хромосом, но в результате удвоения генетического материала и формирования зародыша происходит сбой. При этом только часть клеток плода получит лишнюю хромосому. Доля патологических клеток никогда не превышает 50%. Их число зависит от того, на каком этапе деления начальной клетки произошел сбой. Чем позже это происходит, тем меньше будет доля дефектных клеток. Общее состояние пациента при этом легче, чем при классической форме трисомии 18.
- Частичная трисомия или транслокация – добавление фрагмента третьей хромосомы в результате дефекта деления генетического материала. Транслокационная перестройка ведет к избыточности информации и нарушению генетической последовательности в двух хромосомах. Гены 18 хромосомы переходят с одного участка на другой. Для пациентов с частичной трисомией 18 прогноз лучше, чем для детей с полной формой, но все равно остается неблагоприятным.
Клинически все 3 варианта данного синдрома протекают по одному типу, но первый вариант все же может отличаться более тяжелой формой.
Факторы, способствующие развитию патологии:
- неблагоприятная экология,
- облучение,
- воздействие химикатов и прочих токсинов,
- прием алкоголя беременной женщиной,
- активное и пассивное курение,
- воздействие некоторых лекарств,
- кровное родство супругов,
- заболевания половой сферы,
- возраст матери старше 40 лет.
Вышеперечисленные факторы лишь повышают риск развития данной мутации, а не являются ее непосредственными причинами.
Передача измененного набора хромосом последующим поколениям невозможна. Большинство больных не доживают до репродуктивного возраста. Репродуктивные органы, как и репродуктивные способности у них недоразвиты. Синдром Эдвардса не передается по наследству.
Типы синдрома Эдвардса: как зависит тяжесть проявлений
Исходя из того, какого рода хромосомный дефект имеется при синдроме, выделяют три типа течения:
- Полная трисомия по 18-ой паре.
- Частичная трисомия по 18-ой паре
- Мозаичная форма патологии.
При полной форме (она же относится к классическому синдрому) предполагается, что совершенно во всех клетках имеется дополнительная хромосома. Подобный вариант наиболее тяжелый и встречается наиболее часто, прогнозы при нем самые неблагоприятные.
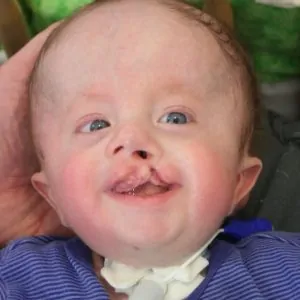 Частичная трисомия относится к редким вариантам патологии, возникает в 3% случаев, при такой проблеме содержится не полная третья хромосома, а только ее кусок. Подобный дефект может образовываться в результате проблем с делением клеток (в особенности их хромосом), что возникает крайне редко. Может быть и вариант того, что лишняя часть 18-ой хромосомы крепится к отдельным молекулам ДНК, приводя к их удлинению и изменению. В таких клетках потом остается две 18-ых хромосомы и часть лишнего материала. Тогда врожденный синдром будет не таким тяжелым, так как в клетках появляется дополнительная информация не всей информации, а только ее части. Прогноз при таком состоянии хотя и неблагоприятный, но течение синдрома гораздо легче, чем при полной форме.
Частичная трисомия относится к редким вариантам патологии, возникает в 3% случаев, при такой проблеме содержится не полная третья хромосома, а только ее кусок. Подобный дефект может образовываться в результате проблем с делением клеток (в особенности их хромосом), что возникает крайне редко. Может быть и вариант того, что лишняя часть 18-ой хромосомы крепится к отдельным молекулам ДНК, приводя к их удлинению и изменению. В таких клетках потом остается две 18-ых хромосомы и часть лишнего материала. Тогда врожденный синдром будет не таким тяжелым, так как в клетках появляется дополнительная информация не всей информации, а только ее части. Прогноз при таком состоянии хотя и неблагоприятный, но течение синдрома гораздо легче, чем при полной форме.
Развитие мозаичной формы проявляется примерно у 5-8% детей с подобной аномалией. Но по механизмам подобный тип синдрома отличается. Интересно, что подобная форма развивается после того, как сперматозоид сливается с яйцеклеткой, и обе получившиеся клетки изначально обладают нормальным набором хромосом – 46ХХ или 46XY. Но при первых же делениях в образованной зиготе образовался сбой и одна из клеток приобрела в силу этого дополнительную хромосому. В результате зародыш приобрел клетки с обычными хромосомами – их 46, другая же часть из них имеет лишнюю хромосому – то есть 47. Количество клеток с лишней хромосомой при таких проблемах не превысит 50%.
Обратите внимание
Чем выше количество дефектных клеток у зародыша и ребенка, тем хуже прогнозы, и зависит их объем от того времени, когда произошел сбой при делении. Чем позже это произошло, тем меньший объем дефектных клеток имеет кроха, и снижено количество проблем развития.
Данная форма получила название мозаичной в силу того, что нормальные клетки перемешаны с дефектными. Атипичные клетки с лишней хромосомой разбросаны хаотично по всему организму, и нет способа, чтобы можно было их каким-либо образом удалить. Эта форма протекает несколько легче, длительность жизни выше.
Диагностика генетической патологии
пренатальная диагностикаПренатальная диагностика синдрома Дауна включает следующие исследования:
- анализ родословной;
- кариотипирование родителей;
- ультразвуковое исследование (УЗИ);
- исследование сывороточных маркеров;
- исследование ДНК плода.
Кариотипирование родителей
лимфоцитымитозтрисомиюПоказанием для проведения данного исследования могут быть:
- возраст матери (во многих странах анализ назначают в обязательном порядке после 35 лет);
- трудности с зачатием ребенка в прошлом (выкидыши, внутриутробная смерть плода, и др.);
- наличие генетических заболеваний в роду у одного из супругов (по результатам анализа родословной);
- место и условия жизни супругов (области с повышенным радиоактивным фоном);
- неблагоприятные условия работы (воздействие сильного электромагнитного излучения, контакт с некоторыми химикатами);
- длительные перебои менструального цикла у женщины и некоторые гормональные заболевания;
- кровное родство с мужем (двоюродный/троюродный брат и т.п.);
- употребление в прошлом наркотических препаратов (оно могло повредить генетический материал в яйцеклетках, что повышает риск, даже если женщина уже много лет как излечилась от зависимости).
Ультразвуковое исследование
В первом триместре беременности при синдроме Дауна на УЗИ можно выявить следующие признаки патологии:
- утолщение воротникового пространства;
- шейная гигрома;
- отсутствие носовой кости;
- отставание плода в росте и весе от нормы на 8 – 10%.
Во втором триместре беременности удается выявить на УЗИ следующие признаки болезни:
- брахицефалия;
- увеличение объема сердечных желудочков;
- кисты в области сосудистых сплетений;
- киста в задней черепной ямке;
- недоразвитие костей лицевого черепа;
- наличие дополнительной складки на шее;
- неправильная форма большой мозговой цистерны;
- непроходимость кишечника (часто в области двенадцатиперстной кишки);
- пороки сердца различной степени тяжести;
- короткие трубчатые кости конечностей;
- аномалии развития пальцев;
- гидронефроз почек.
Исследование сывороточных маркеров
низкое разрешение аппарата, низкая квалификация врача, отсутствие видимых аномалийО наличии синдрома Дауна у плода могут говорить следующие маркеры:
- хорионический гонадотропин человека (ХГЧ);
- плазменный протеин А;
- эстриол;
- альфа-фетопротеин.
ХГЧ
Исследование ДНК плода
представляют собой довольно сложную процедуруК инвазивным методам получения генетического материала плода относятся:
- Кордоцентез. С помощью специальной тонкой иглы делается прокол в передней брюшной стенке матери. Игла вводится в сосуд пупочного канатика, и делается забор крови плода.
- Амниоцентез. Данный метод схож с кордоцентезом, но игла вводится не в сосуды пуповины, а в плодный мешок. Отсюда берут некоторое количество плодной жидкости, которая содержит клетки с поверхности кожи зародыша.
- Биопсия хориона. Техника выполнения аналогична предыдущим исследованиям. С помощью пункции делается забор ворсинок хориона (оболочки плода), которые также содержат ДНК будущего ребенка.
также именуемая ДОТ-тестПодтверждение диагноза после первичного осмотра новорожденного делается по следующим критериям:
- «уплощенное» лицо;
- отсутствие рефлекса Моро (в норме при ударе по поверхности рядом с ребенком он разводит руки в стороны и раскрывает ладони на несколько секунд);
- характерный разрез глаз;
- слабость мышц (мышечная гипотония);
- дефекты развития костей таза;
- повышенная подвижность в суставах конечностей;
- характерное положение мизинца;
- недоразвитие ушных раковин;
- наличие «обезьяньей» складки;
- наличие кожной складки на шее.
После подтверждения диагноза могут быть назначены следующие методы исследования новорожденного:
- УЗИ брюшной полости;
- общий анализ крови и биохимический анализ крови;
- общий анализ мочи и биохимический анализ мочи;
- электрокардиография (ЭКГ);
- эхокардиография (ЭхоКГ);
- рентгенография.
Кроме того, рекомендуется в первые недели или месяцы после рождения пройти осмотр у следующих специалистов:
- оториноларинголог (ЛОР-врач);
- окулист;
- невропатолог;
- кардиолог;
- хирург;
- ортопед.
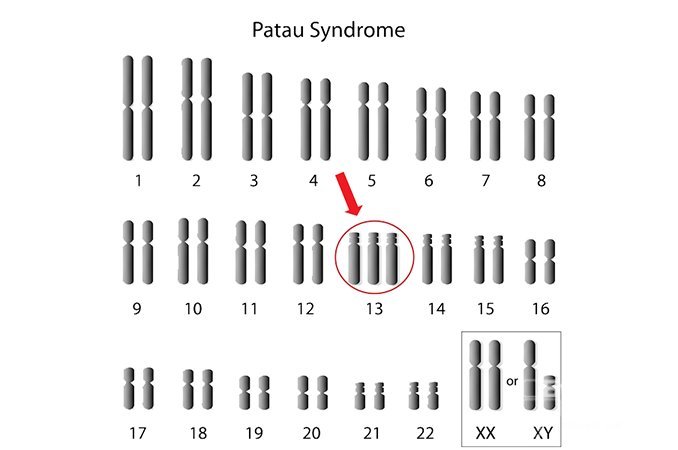
Синдром Патау
Синдром Патау – хромосомное заболевание, обусловленное наличием дополнительной копии 13-ой хромосомы (трисомия по 13-ой хромосоме).
В структуру синдрома Патау входят множественные дефекты нервной системы (микроцефалия, голопрозэнцефалия), глаз (микрофтальмия, катаракта), костно-мышечной системы (полидактилия, расщелины губы и нёба, омфалоцеле), сердца, урогенитальной системы и др.
Для выявления и подтверждения синдрома Патау проводится пренатальный скрининг, исследование кариотипа ребенка после рождения. Детям с синдромом Патау необходима общеукрепляющая терапия; по показаниям – коррекция врожденных пороков развития.
Синдром Патау — хромосомная аномалия, представляющая собой трисомию по 13-ой паре аутосом. Синдрома Патау также встречается в литературе под названиями трисомия D и трисомия 13. Частота рождения детей с синдромом Патау составляет 1:7000-10000; соотношение полов примерно одинаковое.
Клинический симптомокомплекс был описан еще в XVII веке; связь же заболевания с увеличением количества хромосом 13-ой пары была установлена в 1960 г. К. Патау, по имени которого данный синдром и получил свое название.
При синдроме Патау у ребенка имеются множественные и крайне тяжелые аномалии развития, определяющие частые случаи внутриутробной гибели плода и малую продолжительность жизни детей с данной патологией.
Диагностика синдрома Патау
Пренатальная диагностика хромосомных болезней плода (синдрома Патау, синдрома Дауна, синдрома Эдвардса) одинакова. На первом этапе скрининга производится определение биохимических маркеров (бета-ХГЧ, РАРР-А и др.) и УЗИ-исследование, на основании которых рассчитывается риск рождения больного ребенка для данной женщины.
Женщинам, попавшим в группы риска, предлагается проведение инвазивной пренатальной диагностики: биопсии ворсин хориона (8-12 недели), амниоцентеза (14-18 недели) или кордоцентеза (после 20-й недели гестации). В полученных образцах материала плода проводится поиск трисомии по 13-ой хромосоме методом кариотипирования с дифференциальной окраской хромосом или КФ-ПЦР.
Новорожденные с предполагаемым или установленным диагнозом синдрома Патау нуждаются в углубленном комплексном обследовании для выявления тяжелых пороков развития (эхокардиографии, УЗИ органов брюшной полости и почек, нейросонографии, КТ головного мозга и др.). Для определения показаний к оперативному лечению, в первую очередь, необходимы консультации детского кардиохирурга и детского хирурга общего профиля.
Возможности медицинской помощи детям с синдромом Патау ограничены и сводятся, главным образом, к организации хорошего ухода, полноценного питания, профилактике инфекций, общеукрепляющей и симптоматической терапии. Хирургическая помощь может потребоваться для устранения врожденных пороков сердца, расщелин лица и др.
Дети с синдромом Патау находятся под наблюдением педиатра, детского генетика, детского невролога, детского кардиолога, детского офтальмолога, детского травматолога-ортопеда, детского отоларинголога, детского гастроэнтеролога, детского уролога и других специалистов.
Прогноз и профилактика синдрома Патау
В большинстве случаев плод синдромом Патау погибает антенатально или рождается мертвыми. Живорожденные дети также имеют неблагоприятный прогноз для жизни. В большинстве случаев продолжительность их жизни не превышает одного года.
Специфические методы профилактики синдрома Патау не разработаны. При наличии хромосомных заболеваний в предыдущих поколениях или случаев мертворождения перед планированием беременности родителям необходимо пройти медико-генетического консультирование.
Диагностика
Поскольку синдром Эдвардса характеризуется довольно большим количеством ярко выраженных отклонений, его довольно просто диагностировать даже по внешним проявлениям. Однако этого недостаточно, чтобы поставить окончательный диагноз.
Диагностика синдрома Эдвардса складывается из трех этапов — обследование супружеских пар до момента зачатия, беременной женщины до родов и ребенка после появления на свет.
Диагностика до зачатия ребенка — идеальный вариант, но не всегда применимый. Специалисты-генетики могут лишь предположить, каков риск рождения ребенка с хромосомным заболеванием в данной семье.
До момента зачатия врачи собирают семейный анамнез, опрашивая родителей об их родословной.
Большое внимание специалисты уделяют факторам риска: возрасту матери, перенесенным инфекционным заболеваниям, хроническим болезням, вредным привычкам.
Генетический анализ родителей – полноценное исследование, с помощью которого составляется их кариотип и обнаруживаются участки ДНК с дефектными генами.
Диагностика в период внутриутробного развития дает более точные результаты, поскольку обследуют организм плода. Пренатальная диагностика — важный этап в процессе выявления хромосомных нарушений.
- Ультразвуковое исследование плода и допплерография маточно-плацентарного кровотока – неинвазивные методы, полностью безопасные и рекомендованные всем беременным. Признаки синдрома Эдвардса: отставание плода в размерах и массе, большое количество околоплодных вод, видимые аномалии развития черепа и костей, агенезия пупочной артерии, малая величина плаценты, многоводие, брадикардия, отсутствие носовых костей, 2 артерии в пуповине, кисты сосудистых сплетений. Диагностика с помощью ультразвукового исследования является достоверной на 100%.
- Стандартный пренатальный скрининг включает анализ крови на сывороточные маркеры. Полученные результаты соотносят с возрастом беременной женщины и сроком гестации. При отклонении показателей от нормы ставят высокий риск синдрома Эдвардса. В таких случаях показано искусственное прерывание беременности по медицинским показаниям.
- Амниоцентез – клеточный анализ околоплодных вод. Инвазивная методика, осуществляемая путем забора амниотической жидкости шприцем. Ее клетки содержат образцы ДНК плода, которые проверяют на наличие генетических заболеваний.
- Кордоцентез — исследование пупочной крови плода, позволяющее определить генетические аномалии с высокой точностью.
- Биопсия хориона представляет собой пункцию матки через переднюю брюшную стенку и забор ткани для анализа – стандартного генетического исследования.
Беременным, попадающим в группу высокого риска, предлагается проведение инвазивной дородовой диагностики с последующим кариотипированием плода. Инвазивные методы считаются самыми точными и надежными, но требующими оперативного вмешательства и проникновения в оболочку плода. Диагноз подтверждается при помощи определения кариотипа малыша путем КФ-ПЦР.
Диагностика синдрома Эдвардса после рождения самая легкая, быстрая и точная. После выявления некоторых врожденных дефектов проводят генетический анализ для подтверждения диагноза. Основной задачей при рождении ребенка с этой патологией является обнаружение аномалий в развитии внутренних органов, которые обычно приводят к смерти в первые месяцы жизни. Именно на их поиск направлено большинство диагностических процедур непосредственно после рождения. Наиболее важными диагностическими исследованиями, которые должны быть выполнены ребенку с синдромом Эдвардса в первые часы жизни, являются эхокардиография, УЗИ органов брюшной полости и УЗИ почек.