Болезнь шарко
Содержание:
Рекомендации
Можно замедлить прогрессирование заболевания. Это предполагает изменение образа жизни.
Во-первых, требуется регулярно заниматься спортом. Но подходят только те разновидности, где отсутствуют чрезмерные нагрузки. Идеальными вариантами являются езда на велосипеде, пешие прогулки, плавание.
Во-вторых, нельзя допускать физического переутомления, так что и профессия должна быть соответствующей.
В-третьих, требуется удобная обувь. Кроме того, необходимо перейти на правильное, здоровое, сбалансированное питание. Нельзя допускать появления ожирения. При необходимости нужно избавиться от лишнего веса.
Клиническая картина
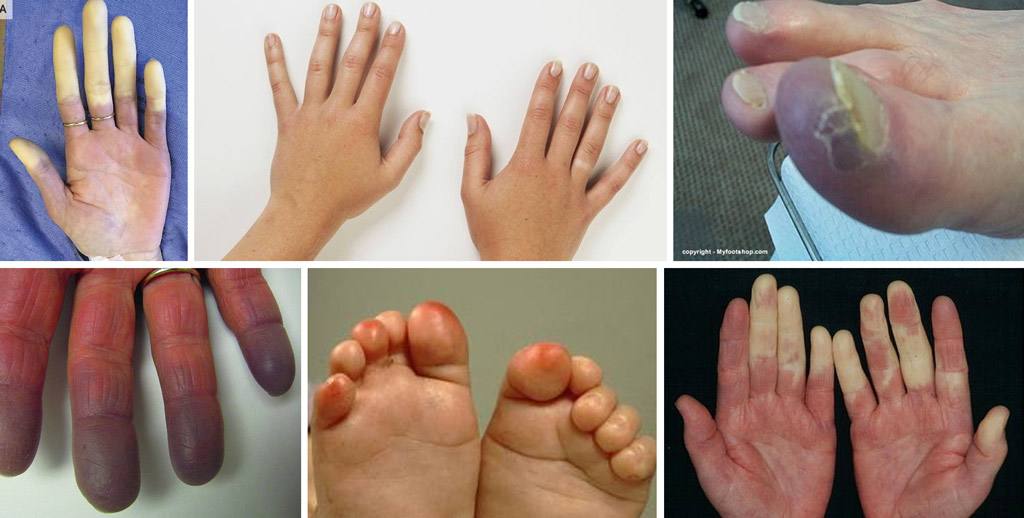
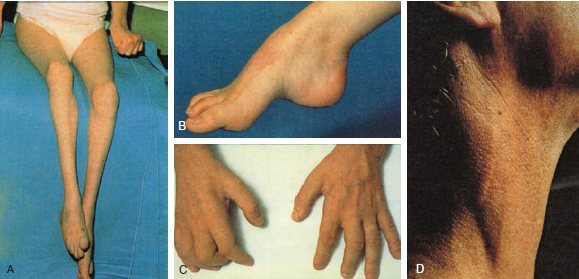
Основным симптомом заболевания являются амиотрофии, которые начинаются симметрично с дистальных отделов нижних конечностей. В первую очередь поражаются разгибатели и абдукторы стопы, в результате чего стопа свисает, появляется характерная походка — степпаж (от англ. steppere — трудовая лошадь). Сгибатели стопы и приводящие мышцы поражаются позже. Атрофия мышц стопы приводит к когтевидной установке пальцев и деформации стопы, напоминающей стопу Фридрейха. Амиотрофический процесс постепенно распространяется на более проксимальные отделы. Однако в подавляющем большинстве случаев проксимальные отделы конечностей остаются сохранными; процесс не распространяется также на мышцы туловища, шеи и головы. При атрофии всех мышц голени образуется болтающаяся стопа. На этой стадии болезни часто отмечается симптом «топтания», когда больные в положении стоя постоянно переминаются с ноги на ногу. Атрофия мышц может распространяться на нижнюю часть бедер. Форма ноги в этих случаях напоминает опрокинутую бутылку. Как правило, через несколько лет атрофии распространяются и на верхние конечности. В первую очередь поражаются мелкие мышцы кисти, в результате чего кисть приобретает форму «обезьяньей лапы». Затем в процесс вовлекаются мышцы предплечья. Мышцы плеча страдают в значительно меньшей степени
Обращает на себя внимание то, что, несмотря на выраженные атрофии мышц, больные могут в течение длительного времени сохранять трудоспособность. При неавральной амиотрофии часто наблюдаются нередко выраженные фасцикулярные подергивания в мышцах конечностей. При электромиографическом исследовании выявляются признаки невритического, переднерогового и супрасегментарного типов нарушения мышечного электрогенеза
При электромиографическом исследовании выявляются признаки невритического, переднерогового и супрасегментарного типов нарушения мышечного электрогенеза.
Мужчины болеют несколько чаще женщин. Заболевание обычно начинается в детском возрасте — во второй половине первого или в первой половине второго десятилетия жизни. Однако возраст начала болезни может широко колебаться в разных семьях, что допускает возможность генетической гетерогенности данного заболевания.
Течение болезни — медленно прогрессирующее. Между началом амиотрофии в верхних и нижних конечностях может проходить до 10 лет и больше. Иногда процесс обостряется в связи с различными экзогенными вредностями. В отдельных случаях в течение длительного времени состояние больных может оставаться стационарным.
Для невральной амиотрофии Шарко-Мари характерны также нарушения чувствительности. В дистальных отделах конечностей определяется гипестезия, причем поверхностные виды чувствительности, главным образом болевая и температурная, страдают в значительно большей степени. Могут отмечаться боли в конечностях, повышенная чувствительность к давлению нервных стволов.
В ряде случаев имеют место трофические нарушения — отек и цианоз кожных покровов конечностей.
Клинические проявления заболевания в ряде семей могут варьировать. Описаны семьи, где наряду с типичной невральной амиотрофией имели место случаи гипертрофического полиневрита. В связи с этим некоторые авторы объединяют эти заболевания в одну нозологическую форму.
Неоднократно подчеркивалась связь между невральной амиотрофией и наследственной атаксией Фридрейха. Наблюдались семьи, у одних членов которых имелась невральная амиотрофия, у других — атаксия Фридрейха. Описаны промежуточные формы между этими заболеваниями; у отдельных больных типичная клиническая картина атаксии Фридрейха через много лет сменялась картиной невральной амиотрофии, которую отдельные авторы считают даже промежуточной формой между атаксией Фридрейха и нейрофиброматозами.
Процедуры и операции
Особое внимание уделяется немедикаментозным методам терапии. Комплекс мероприятий, позволяющих достичь максимального терапевтического эффекта:
- Лечебная физическая культура. Регулярные занятия ЛФК повышают мышечный тонус. Наибольший эффект достигается при достижении пассивных упражнений (занятия вместе со специалистом) и активных (выполняются самостоятельно).
- Электростимуляция. Направленная подача электрических импульсов улучшает проводимость по периферической нервной системе, активирует метаболические процессы в паретичных мышцах, усиливает нейротрофику.
- Бальнеотерапия. Грязевые аппликации и грязевые ванны замедляют формирование контрактур и улучшают работу вегетативной нервной системы.
- Массаж. Различные виды массажа (аппаратный и ручной) улучшают лимфатический отток и нормализуют кровообращение. Рекомендован расслабляющий, стимулирующий и вибрационный массаж.
- Ортопедическая терапия. Ношение специальной ортопедической обуви позволяет предотвратить развитие грубой деформации. Из-за мышечной слабости развивается нестабильность суставов. Подтяжки, ортезы и другие специальные приспособления используются для фиксации стоп в необходимом положении.
При комплексном подходе к терапии повышается уровень мышечной силы, нормализуется походка и исправляются различные нарушения равновесия. Грамотно подобранное лечение повышает социальную и бытовую адаптацию, восстанавливает работоспособность пациента.
Факторы риска и причины ШMT
ШMT является наследственным заболеванием, так что люди, которые имеют близких родственников с заболеванием, имеют более высокий риск развития болезни.
Заболевание поражает периферические нервы. Периферический нервы состоит из двух основных частей: аксона — внутренняя часть нерва и миелиновой оболочки, которая является защитным слоем вокруг аксона. ШМТ может влиять на аксон и миелиновую оболочку.
При ШMT 1
мутируют гены, которые вызывают распад миелиновой оболочки. В конце концов, повреждается аксон, и мышцы пациента больше не получают четких сообщений от мозга. Это приводит к мышечной слабости и потере чувствительности или онемению.
При ШМТ 2
мутирующий ген влияет непосредственно на аксоны. Сигналы передаются не достаточно сильно, чтобы активизировать мышцы и органы чувств, так что пациенты имеют слабые мышцы, плохую чувствительность или онемение.
ШМТ 3
илиболезнь Дежерин-Соттас , редкий тип заболевания. Повреждение миелиновой оболочки приводит к выраженной мышечной слабости и чувствительности. Симптомы могут быть заметны у детей.
ШМТ 4
является редким заболеванием, которое влияет на миелиновую оболочку. Симптомы обычно появляются в детстве, и пациенты часто нуждаются в инвалидном кресле.
ШМТ Х
вызывается мутацией Х-хромосомы. Она чаще встречается у мужчин. Женщина с CMT X будет иметь очень слабые симптомы.
Причины
Генетические заболевания определяются комбинацией генов для конкретного признака, которые находятся на хромосомах, полученных от отца и матери.
Человек получивший один нормальный и один ген заболевания, является носителем, но обычно не проявляет симптомов.
- Риск для двух родителей-носителей передачи дефектного гена детям – 25%.
- Иметь ребенка – носителя -50%.
- Шанс для ребенка получить нормальные гены – 25%.
Риск одинаковый для мужчин и женщин.
Доминантные генетические расстройства возникают, когда для появления болезни необходима только одна копия аномального гена. Аномальный ген может быть унаследован от любого из родителей или быть результатом новой мутации (изменения гена).
Риск передачи ненормального гена от пострадавшего родителя к потомству составляет 50% для каждой беременности независимо от пола ребенка.
Х-сцепленные доминантные генетические нарушения вызваны аномальным геном на Х-хромосоме. Мужчины с аномальным геном страдают более сильно, чем женщины.
Наследственная нейропатия подразделяется на несколько типов, называемых CMT1, CMT2, CMT3, CMT4 и CMTX.
CMT1
Является доминирующей формой расстройства, при котором скорости проводимости нерва являются медленными. Более распространен, чем CMT2. Вызван аномальными генами, которые участвуют в структуре и функции миелина. Дополнительно подразделен на CMT1A, CMT1B, CMT1C, CMT1D, CMT1X на основе специфических аномалий.
- CMT1A появляется из-за дублирования гена PMP22, который расположен на хромосоме 17 при 17p11.2. Является наиболее распространенным типом.
- CMT1B вызван аномалиями в гене MPZ на хромосоме 1 при 1q22.
- CMT1C появляется от аномалий SIMPLE, расположенном на 16 хромосоме при 16p13.1-p12.3.
- CMT1D аномалия EGR2, расположенной на 10, при 10q21.1-q22.1.
- CMT1X возникает от мутаций GJB1 (Xq13.1), Он кодирует белок связного соединения connexin32.
Узнать больше Что такое синдром Шихана?
CMT2
Является аутосомно-доминирующей формой расстройства, при котором скорости проводимости нерва обычно нормальны или немного медленнее, чем обычно. Вызван аномальными генами, участвующими в структуре и функции аксонов. Дополнительно подразделен на CMT2A-2L на основе мутаций.
- CMT2A, является наиболее распространенным и обусловлен ошибками MFN2, расположенным на хромосоме 1, в 1p36.2.
- CMT2B от мутаций RAB7 на хромосоме 3 при 3q21.
- CMT2C вызывается неизвестным геном на 12 – 12q23-34.
- CMT2D ошибки GARS, на 7 – 7p15.
- CMT2E от NEFL, расположенном на 8 – 8p21.
- CMT2F ошибки гена HSPB1.
- CMT2L мутации HSPB8.
Доминирующая промежуточная DI-CMT. Она названа так из-за «промежуточной» скорости проводимости, неопределенности относительно того, является ли нейропатия аксональной или демиелинизирующей. Известно, что доминантные мутации в DMN2 и YARS вызывают этот фенотип.
CMT3
Также называемый болезнью Дежерин-Соттас, у индивидуумов с этим расстройством обнаружена мутация в одном из генов, ответственных за CMT1A, CMT1B, CMT1D, CMT4.
CMT4
Аутосомно-рецессивная форма состояния. Он подразделяется на CMT4A, CMT4B1, CMT4B2, CMT4C, CMT4D, CMT4E, CMT4F.
- CMT4A вызван аномалиями GDAP1. Ген находится на хромосоме 8 при 8q13-q21.
- CMT4B1 – аномалия MTMR2 на 11 – 11q22.
- CMT4B2 от аномалий SBF2 / MTMR13, на 11 при 11p15.
- CMT4C ошибки KIAA1985, на хромосоме 5 – 5q32.
- CMT4D мутации NDRG1, на хромосоме 8 – 8q24.3.
- CMT4E, также известная как врожденная гипомиелиническая невропатия. Происходит от аномалий EGR2, на 10 – 10q21.1-q22.1.
- CMT4F аномалии PRX, на хромосоме 19 – 19q13.1-q13.2.
- CMT4H ошибки FDG4.
- CMT4J мутации FIG4.
Однако большинство случаев CMT2 не вызваны мутациями этих белков, поэтому многие генетические причины еще не обнаружены.
CMTX
Является X-связанной доминирующей формой расстройства. На CMT1X приходится около 90% случаев. Конкретный белок, ответственный за оставшиеся 10% CMTX, еще не идентифицирован.
Аутосомно-рецессивный CMT2 происходит из-за мутаций LMNA, GDAP1.
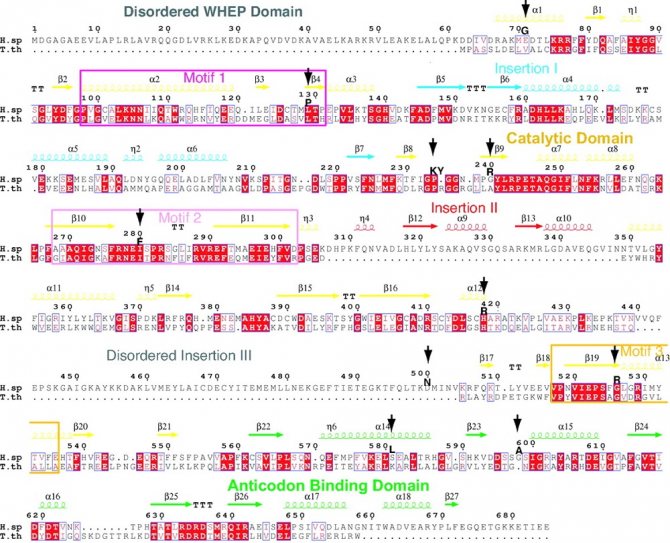
Структурное выравнивание с элементами вторичной структуры, расположенными сверху. Десять мутаций, вызывающих CMT, отмечены вертикальными стрелками. (Нажмите, чтобы увеличить)
Историческая справка
Впервые официальное описание заболевания появилось в 1886 году в журнале La revue de medicine. Одним из авторов был Жан Мартен Шарко (Франция), которому на тот момент уже шел 61 год и он был известным медиком страны. Вторым автором, участвующим в описании заболевания и его симптомов, был его ученик, который практиковал всего лишь 3 года и звали его Пьер Мари.
Вторая статья была написана совсем молодым неврологом Говардом Генри Тутом (Англия), он только год назад получил приставку к своему имени – MD. И с того момента, заболевание получило название, состоящее из фамилий трех врачей.
Понятное дело, что это были не первые публикации о перонеальной мышечной атрофии. Еще ранее в записях Рудольфа Вирхова (1855 год) присутствует описание заболевания, но не столь подробное и без признаков.
Лечение
Терапия пациентов с болезнью Шато проводится в стационарных условиях. Какого-либо специфического лечения нет, чтобы замедлить прогрессирование демиелинизации и аксональной дегенерации. Индивидуальная, грамотно подобранная терапия позволяет улучшить качество жизни пациента.
Доктора
специализация: Невролог / Ортопед

Крутых Ирина Валерьевна
5 отзывовЗаписаться
Подобрать врача и записаться на прием
Баклофен
Прозерин
Колекальциферол
Галантамин
Медикаменты, которые применяются при невральной амиотрофии ШМТ:
Витамины. Витаминные комплексы позволяют восстановить структуру нервных волокон, улучшить микроциркуляцию
С особой осторожностью назначают витамин В6 из-за его нейротоксичности. Проведённые исследования показали, что аскорбиновая кислота способна подавлять синтез РМР22 периферического белка миелина.
Витамин Д и кальций
У 40% пациентов диагностируется остеопороз, что требует назначения Колекальциферола и препаратов кальция для профилактики переломов.
Миорелаксанты. Для уменьшения выраженности болевого синдрома от мышечных сокращений назначают препараты, которые расслабляют скелетную мускулатуру (Толперизон, Баклофен).
Антихолинэстеразные препараты. Для улучшения нервно-мышечной проводимости при ШМТ 2 типа назначают Галантамин, Прозерин.
1.2.2. Невральная амиотрофия шарко-мари-тута.
Частота 1:500000 населения. Наследуется по аутосомно-доминантному, аутосомно-рецессивному сцепленному с Х-хромосомой типу. Обнаруживается сегментарная демиелинизация в нервах, в мышцах — денервация с явлениями «пучковой» атрофии мышечных волокон.
КЛИНИКА. Первые признаки заболевания чаще проявляются в 15 — 30 лет, реже в дошкольном возрасте. Характерными симптомами являются мышечная слабость, патологическая утомляемость в дистальных отделах ног. Больные быстро устают при длительном стоянии на одном месте и нередко для уменьшения утомления в мышцах прибегают к ходьбе на месте («симптом топтания«). Реже заболевание начинается с чувствительных расстройств — болей, парестезий, ощущения ползания мурашек. Атрофии первоначально развиваются в мышцах голеней и стоп. Мышечные атрофии, как правило, симметричные. Поражается перонеальная группа мышц и передняя большеберцовая мышца. Вследствие атрофий ноги резко сужаются в дистальных отделах и приобретают форму «перевернутых бутылок» или «ног аиста». Стопы деформируются, становятся «выеденными» с высоким сводом. Парез стоп изменяет походку больных. Они ходят, высоко поднимая ноги; ходьба на пятках невозможна. Атрофии в дистальных отделах рук — мышцах тенара, гипотенара, а также в мелких мышцах кистей присоединяются позже. Ахилловы рефлексы снижаются в ранних стадиях болезни, а коленный рефлекс, рефлекс с трех-двуглавой мышц плеча длительное время остаются сохранными. Чувствительные расстройства объективно определяются нарушениями поверхностной чувствительности по периферическому типу (тип «перчаток» и «носков»). Часто имеются вегетативно-трофические нарушения — гипергидроз стоп и кистей, гиперемия кистей и стоп. Интеллект обычно сохранен.
Течение медленно прогрессирующее. Прогноз благоприятен.
Диагностика. Тип наследования, атрофии дистальных отделов конечностей, расстройства чувствительности по полиневритическому типу, медленное прогрессирующее течение, результаты электромиографии (снижение скорости проведения по периферическим нервам), биопсия нервов.
ЛЕЧЕНИЕ ПМД. Применяют витамины группы В, С Е, а также АТФ, церебролизин, ноотропил, энцефабол, фосфаден, карнитина хлорид, метионин, лецитин, глутаминовая кислота, ретаболил. Положительный эффект дают антихолинэстеразные препараты (прозерин, местинон, галантамин). Показаны средства, улучшающие микроциркуляцию: никотиновая кислота, трентал, пармидин. Наряду с медикаментозной терапией применяют ЛФК, массаж, электрофорез лекарственных средств (прозерин, кальция хлорид), диадинамические токи, синусоидальные модулированные токи, электростимуляция, ультразвук, озокерит, грязевые аппликации, радоновые, хвойные, сульфидные и сероводородные ванны, оксигенобаротерапия. Показано ортопедическое лечение при контрактурах конечностей, умеренной деформации позвоночника и асимметричном укорочении конечностей.
13.2. Семейная атаксия фридрейха.
Наследственное дегенеративное заболевание нервной системы, характеризующееся синдромом поражения задних и боковых столбов спинного мозга. Тип наследования аутосомно-рецессивный с неполной пенетрантностью патологического гена. Мужчины и женщины болеют одинаково часто.
КЛИНИКА. Начало заболевания относится к 6 — 15-летнему возрасту. Первым симптомом болезни является неустойчивая походка. В ранних стадиях атаксия выражена преимущественно в ногах. По мере прогрессирования заболевания нарушения координации распространяются на руки и лицо. При неврологическом обследовании выявляются крупноразмашистый нистагм, атаксия в руках и ногах, адиадохокинез, дисметрия, скандированная речь, расстройства мышечно-суставного чувства и вибрационной чувствительности. Нарушается почерк. Ранним симптомом является снижение, а затем угасание сухожильных и периостальных рефлексов. Мышечный тонус понижен. В более поздних стадиях болезни присоединяются афферентный парез нижних, а затем верхних конечностей, нередко патологические пирамидные рефлексы, дистальные мышечные атрофии. Интеллект снижен. Заболевание медленно прогрессирует. Средняя продолжительность жизни 10 — 15 лет с момента его развития.
Диагностика. Заболевание распознается на основании характерных симптомов — деформаций стоп по типу стопы Фридрейха (высокий свод, экстензия основных фаланг пальцев стопы, флексия концевых фаланг), позвоночника, поражения миокарда, эндокринных расстройств.
ЛЕЧЕНИЕ. Применяются симптоматические средства: общеукрепляющие препараты, ЛФК, массаж. В некоторых случаях производится хирургическая коррекция деформации стоп.
Болезнь Шарко лечение
Для основного лечения болезни Шарко важно замедлить прогрессирующий процесс заболевания и продлить период, при котором больной сохранит способность самообслуживания. А также необходимо уменьшить характерные выраженные симптомы и поддержать как можно дольше стабильный уровень жизни больного
Госпитализации подлежат больные в том случае, если необходимо провести первичное обследование, для точного диагностирования болезни, или проведения гастротомии.
На данный момент разработан только один препарат, который существенно замедляет процессы прогрессирования болезни Шарко – это Рилузол. Применение этого препарата продлевает жизнь больным в среднем на три месяца. Его назначают строго по показаниям, после подтверждения диагноза, но исключив при этом другие причины поражения мотойнеров, а также если для болезни Шарко характерна длительность не менее пяти лет, нет в наличии трахеостомии и с форсированной жизненной ёмкостью лёгких в 60%. Назначают Ризул пожизненно по 100 мг в сутки, с постоянным контролем печёночных трансаминаз через каждые три месяца, чтобы избежать присоединения гепатита лекарственной этиологии.
Проведение патогенетического лечения болезни Шарко с применением Ксалипродена, противопаркинсонских средств, иммуномодуляторов, антиоксидантов, Церебролизина оказалось неэффективным. Поэтому паллиативная терапия пытается приостановить прогрессирование депрессий, дисфагий, фасцикуляций, спастичности и дизартрии. Чтобы улучшить метаболизм мышц назначают курсами Карнитин, Левокарнитин, Креатин на два месяца по три раза в год. Для того чтобы пациентам было легче ходить, врачи рекомендуют носить только ортопедическую обувь, а при необходимости использовать трость и ходунки.
Назначение гастротомии улучшает состояние больного и продлевает ему жизнь. А вот трахеостомия и искусственная вентиляция лёгких – это приговор и последний этап заболевания. Как правило, они сигнализируют о скором летальном исходе.
Прогноз при болезни Шарко всегда неблагоприятный. Продолжительность жизни при пояснично-крестцовой форме – до трёх лет, при бульбарной – до четырёх, а в основном с этим заболеванием пациенты не живут более пяти лет.
Как проявляется болезнь Шарко-Мари-Тута?
Болезнь Шарко-Мари-Тута проявляется даже в пределах одной семьи не всегда одинаково. И дело не в многообразии её признаков. А в том, что гены, кодирующие данную патологию, способны с разной степенью выраженности формировать симптомы. Проще говоря, имея идентичную «поломку» в хромосомах, признаки недуга у отца и сына будут иметь индивидуальную окраску.
Общие симптомы
Клиническая картина заболевания практически не зависит от его типа и включает:
- атрофию мышц дистальных, то есть наиболее отдалённых от туловища, отделов конечностей;
- снижение сухожильных и периостальных рефлексов;
- изменение чувствительности, характеризующееся её выпадением, но никогда не сопровождающееся появлением ощущения покалывания или «ползания мурашек»;
- деформацию опорно-двигательного аппарата – сколиоз, увеличение свода стопы и др.
Но всё же существует ряд признаков несколько отличающих течение заболевания при различных его вариантах.
Первый тип
Болезнь Шарко-Мари-Тута первого типа нередко протекает в исключительно стёртой форме, при которой пациенты не ощущают изменений в организме и не обращаются за медицинской помощью вообще. Если же патология себя проявила, то происходит это в период первого, максимум второго, десятилетия жизни.
При этом наблюдаются:
- болезненные судороги в мышечном массиве голени, причём редко в икроножной мышце, чаще в передней группе мышц. Подобные спазмы нарастают после периода длительной физической нагрузки (ходьбы, занятий спортом, езде на велосипеде);
- изменения в походке, связанные с постепенным усилением слабости в мышцах. При этом у детей это может дебютировать в хождении на цыпочках;
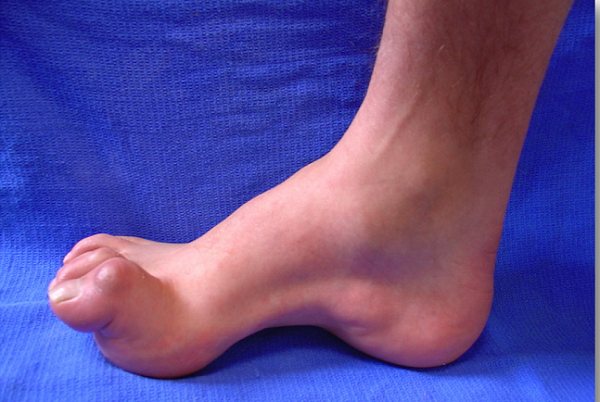
- деформация стоп с формированием высокого свода последних и наличием молоточкообразных пальцев, которая развивается как результат дисбаланса в тонусе сгибателей и разгибателей;
- атрофия мышц, начинающаяся со стоп и поднимающаяся на голень. Затем процесс затрагивает кисть – появляется тремор в руках и выраженная слабость в пальцах, особенно при попытке выполнять мелкие движения. К примеру, застёгивать пуговицы, перебирать крупу;
- угнетение или полное отсутствие сухожильных и периостальных рефлексов, а именно ахиллового, карпорадиального, при сохранных с более проксимальных отделов рук и ног. То есть коленный и рефлексы с двуглавой и трёхглавой мышцы остаются интактными;
- нарушения чувствительности в кистях и стопах, выражающиеся в её постепенном выпадении. Причём стартует патология с вибрационной и тактильной сферы, распространяясь на суставно-мышечные и болевые ощущения;
- сколиоз и кифосколиоз;
- утолщение нервных стволов, чаще всего поверхностного малоберцового и большого ушного.
Для болезни Шарко характерна атрофия мышц именно в дистальных отделах конечностей. При этом если не выражена подкожно-жировая клетчатка, объём голени и бедра разительно отличается, и ноги приобретают вид таковых у аиста или походят на перевёрнутую бутылку для шампанского.
Невральная амиотрофия Шарко Мари первого типа имеет атипичные формы. Одна из них — синдром Руси-Леви, при котором наблюдается выраженный тремор при попытке удержать руки в одном положении и неустойчивость при ходьбе. Сюда же относится заболевание, проявляющееся, помимо стандартных симптомов, парезами, гипертрофией мышц голени, резким выпадением чувствительности и ночными судорогами в икроножных мышцах.
Второй тип
Для болезни Шарко-Мари-Тута второго типа, кроме более позднего начала, характерны:
- менее выраженные изменения чувствительности;
- более редкая встречаемость деформаций стопы и пальцев;
- наличие синдрома беспокойных ног (возникают неприятные ощущения в ногах во время отхода ко сну, заставляющие пациента двигаться, что облегчает состояние);
- нередко сохранная сила в кисти;
- отсутствие утолщения нервных стволов.
При синдроме Шарко-Мари-Тута, передающемся через Х-хромосому, могут встречаться нейросенсорная тугоухость (снижение слуха) и транзиторная энцефалопатия, возникающая после физической нагрузки на высоте. Для последней характерно появление симптомов через 2-3 дня после занятий. Признаками патологии становятся шаткость, нарушение речи, глотания, слабость в проксимальных отделах рук и ног. Обычно клиническая картина недуга исчезает самостоятельно в течение пары недель.