Все о болезни вильсона-коновалова
Содержание:
- Клиническая картина и течение
- Причины болезни Вильсона
- Патологическая анатомия
- Течение
- Какие изменения мозга вызывают признаки болезни?
- Токсическое действие меди при болезни Вильсона
- Общая картина заболевания
- Лечение Болезни Вильсона-Коновалова (гепатоцеребральной дистрофии):
- Терапия патологии
- Определение болезни. Причины заболевания
- Симптомы болезни Вильсона
- Лечение
- Лечение
- Обратите внимание
- Выводы
Клиническая картина и течение
Гепато-церебральная дистрофия начинается в детском или молодом возрасте и имеет хроническое прогрессирующее течение. Во многих случаях появлению симптомов поражения нервной системы предшествуют висцеральные расстройства в виде нарушения деятельности печени и желудочно-кишечных расстройств (желтуха, боли в правом подреберье, диспептические явления). Порой развивается выраженный гепато-лиенальный синдром. Со стороны нервной системы на первый план выступают экстрапирамидные симптомы в виде мышечной ригидности, гиперкинезов и расстройств психики. Пирамидные симптомы могут быть, но чаще отсутствуют. Чувствительность обычно не нарушена.
Типичным симптомом болезни являются кольца Кайзера-Флейшера — отложения по периферии роговой оболочки содержащего медь зеленовато-бурого пигмента, более выраженные на поздних стадиях. Иногда отмечается желтовато-коричневая пигментация кожи туловища и лица. Часты геморрагические явления (кровоточивость дёсен, носовые кровотечения, положительная проба жгута), мраморность кожи, акроцианоз. Капилляроскопия обнаруживает атонию капилляров и застойность кровотока. Отмечаются суставные боли, профузные поты, остеопороз, ломкость костей.
Патология печени клинически выявляется примерно у 30 % больных, а в ряде случаев она может быть обнаружена только функциональными пробами, например пробой с нагрузкой галактозой, пробой Квинка, пробой Бергмана-Эльботта, бромсульфофталеиновой пробой; количество билирубина в крови и уробилина в моче обычно увеличено; изменены осадочные реакции Таката-Ара и Грея, обычны лейкопения, тромбоцитопения, гипохромная анемия.
Различают 5 форм гепато-церебральной дистрофии:[уточнить]
- Брюшная форма — тяжёлое поражение печени, приводящее к смерти раньше появления симптомов со стороны нервной системы; заболевают дети. Её продолжительность от нескольких месяцев до 3-5 лет.
- Ригидно-аритмогиперкинетическая, или ранняя форма — отличается быстрым течением; начинается также в детском возрасте. В клинической картине преобладают мышечная ригидность, приводящая к контрактурам, бедность и замедленность движений, хореоатетоидные или торсионные насильственные движения. Характерны дизартрия и дисфагия, судорожный смех и плач, аффективные расстройства и умеренное снижение интеллекта. Заболевание длится 2-3 года, заканчивается летально.
- Дрожательно-ригидная форма встречается чаще других; начинается в юношеском возрасте, течёт медленнее, порой с ремиссиями и внезапными ухудшениями, сопровождающимися субфебрильной температурой; характеризуется одновременным развитием тяжёлой ригидности и дрожания, дрожание очень ритмичное (2-8 дрожаний в секунду), резко усиливается при статическом напряжении мышц, движениях и волнении, в покое и во сне исчезает. Иногда обнаруживаются атетоидные хореоформные насильственные движения; наблюдаются также дисфагия и дизартрия. Средняя продолжительность жизни около шести лет.
- Дрожательная форма начинается в возрасте 20-30 лет, течёт довольно медленно(10-15 лет и больше); дрожание резко преобладает, ригидность появляется лишь в конце болезни, а порой наблюдается гипотония мышц; отмечается амимия, медленная монотонная речь, тяжёлые изменения психики, часты аффективные вспышки. Наблюдаются эпилептиформные припадки.
- Экстрапирамидно-корковая форма встречается реже других форм. Типичные для гепато-церебральной дистрофии нарушения в дальнейшем осложняются апоплектиформно развивающимися пирамидными парезами, эпилептиформными припадками и тяжёлым слабоумием (обнаруживаются обширные размягчения в коре больших полушарий). Длится 6-8 лет, заканчивается летально.
Наибольшая летальность (50 %) отмечается при печёночной форме с массивным некрозом и гемолизом у детей до 6 лет. Смерть больных от неврологических нарушений при отсутствии лечения наступает через 5-14 лет. Основная причина при этом интеркуррентные заболевания или желудочно-кишечные кровотечения, портальная гипертензия.
Причины болезни Вильсона
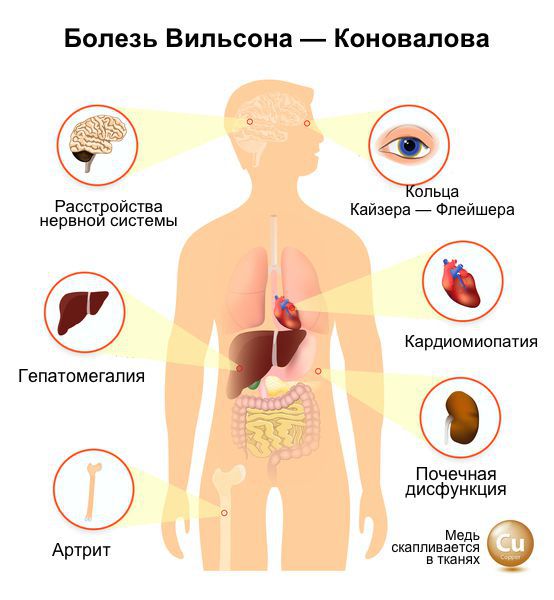
Болезнь Вильсона — наследственное, поддающееся лечению заболевание, при котором происходит постепенное накопление меди в печени, головном мозге (преимущественно в базальных ядрах), роговице, почках, что вызывает тяжелые функциональные нарушения, ведущие к необратимым повреждениям. Без лечения заболевание заканчивается смертью, однако своевременная постановка диагноза и лечение устраняют или предотвращают его симптомы.
Болезнь Вильсона наследуется аутосомно-рецессивно. Распространенность гетерозигот по мутантному гену составляет 1:200, а гомозигот — 1:30 000. Ген, ответственный за болезнь Вильсона, расположен на 13-й хромосоме вблизи гена, кодирующего эстеразу D. У 95% больных наблюдается дефицит или полное отсутствие церулоплазмина (сывороточный белок, играющий главную роль в транспорте меди). Это обусловлено снижением транскрипции гена церулоплазмина, расположенного на 3-й хромосоме.
Медь (Cu) — важнейший микроэлемент, который входит в состав ферментов, например цитохромоксидазы, тирозиназы, супероксиддисмутазы и др.
Метаболизм меди. Поступление Cu в организм в норме составляет приблизительно 2-5 мг в сутки, из которых 40-60 % всасываются в желудке и верхнем отделе двенадцатиперстной кишки. С помощью переносчика Си поступает в клетки печени, связывается с белками или включается в состав церулоплазмина (ферроксидаза), который относительно прочно связывает шесть атомов Cu. В комплексе с церулоплазмином Cu выделяется в плазму (приблизительно 93 % от содержания в плазме), где она окисляет Fe2+ в Fe3+. Незначительное количество Cu, связанное с церулоплазмином, высвобождается в периферических тканях. Экскреция Cu в желчь осуществляется с помощью АТФазы Р-типа, которая называется Cu-АТФаза (АТР7В). «Состарившийся», не содержащий сиаловых кислот церулоплазмин разрушается в печени, Си высвобождается, прочно связывается с белками желчи и выводится из организма с калом. В сутки выделяется приблизительно 1,2 мг меди.
Болезнь Вильсона (гепатолентикулярная дегенерация) — аутосомно-рецессивное нарушение метаболизма Cu, при котором в печени, ЦНС, глазах и других органах происходит ее избыточное отложение. Заболевание вызывается мутациями гена АТР7В, кодирующего белок CU-ATP7B. Мутация приводит к снижению выведения значительного количества Cu с желчью и уменьшению включения Cu в церулоплазмин. В результате при общей концентрации Си ниже нормы в печени и затем в плазме накапливается свободная или слабо связанная медь. В такой форме Cu токсична, поскольку связывается преимущественно с сульфгидрильными группами белков и способствует образованию свободных радикалов O2 и перекисному окислению липидов.
Накопление свободной Cu вызывает развитие анемии и хронического гепатита, который впоследствии приводит к циррозу. При молниеносном гепатите из некротически измененной ткани печени внезапно высвобождается большое количество Cu, что может вызвать гемолитический криз. Накопление Cu в ЦНС приводит к многочисленным и разнообразным неврологическим, нервно-мышечным и психогенным расстройствам. Отложение Cu в виде зернистой массы в десцеметовой оболочке глаз способствует образованию кольца Кайзера—Флейшера по периферии роговицы. В патологический процесс могут вовлекаться почки, скелет и сердце. Поскольку избыточное отложение Си обусловлено мутацией гена АТР7В, излечение от болезни Вильсона возможно путем трансплантации печени.
Патологическая анатомия
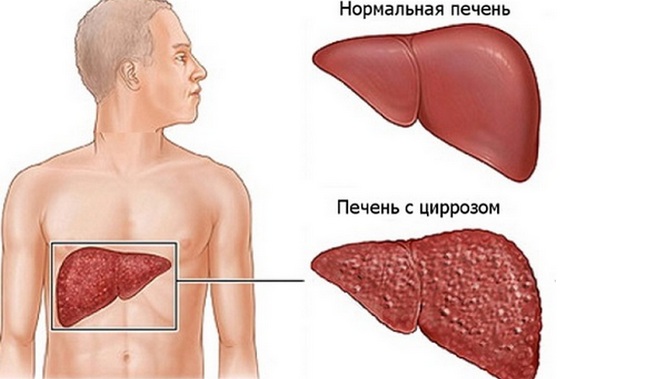
Печень уменьшена и бугриста. Атрофический цирроз печени мало отличается от лаэннековского. Микроскопически участки печени нормального строения, чередуются с некротическими. Отмечаются и регенеративные явления, разрастание рубцовой и молодой соединительной ткани, богатой сосудами. В печени содержится большое количество зёрен меди. Изменения сосудов печени имеют большое значение в нарушении барьерной функции, так как кровь воротной вены может, минуя печень, может поступать прямо в нижнюю полую вену. Наблюдается увеличение селезёнки, гиперплазия её пульпы, явления нефрита, жировое перерождение почечного эпителия, отложение извести.
Изменения в нервной системе расцениваются как ангиотоксические и цитотоксические; поражение мозга не является системным и не ограничивается подкорковыми узлами. Процесс распространяется на различные отделы нервной системы, но изменения в области лентикулярных ядер являются наиболее выраженными и постоянными.
Поражение других отделов имеет следующую последовательность: хвостатое тело, наружный членик бледного шара, глубокие слои мозговой коры, зубчатые ядра мозжечка, ядра гипоталамической области. Остальные отделы мозга поражаются меньше.
Ангиотоксические изменения стоят на первом плане. Они выражаются атонией сосудов, особенно мелких, их расширением, переполнением кровью, образованием стазов, но без развития тромбозов. В различных отделах мозга наблюдаются кровоизлияния. Отмечаются также изменения стенок сосудов с размножением клеток адвентиции и эндотелия, отложением липоидов, гиалинозом капилляров. Вследствие повышенной проницаемости сосудов развивается периваскулярный отёк и аноксическое разрушение нервной ткани. Богатые капиллярами участки мозга поражаются больше всего. Распространение этих изменений в головном мозгу иногда очень обширно, а их характер лучше всего согласуется с представлением о токсическом воздействии крови на сосудистую стенку.
Цитологические изменения выражаются дегенерацией ганглиозных нервных и макроглиальных клеток. Для гепатолентикулярной дегенерации характерно изменение глии (глия Альцгеймера). Глия Альцгеймера образуется из обычных астроцитов, которые бывают двух типов. К первому типу относятся громадные глиозные клетки с бледной протоплазмой и большим, богатым хроматином ядром. Клетки со сморщенным, пикнотическим ядром называют клетками Опальского. Ко второму типу клеток Альцгеймера относятся гигантские ядра, лишенные протоплазмы, которые на окрашенных тионином препаратах представляются «голыми».
Клетки Альцгеймера встречаются в различных отделах мозга, чаще всего в подкорковых узлах, зрительном бугре, гипоталамической области, зубчатом ядре мозжечка. Ганглиозные нервные клетки подвергаются такого же рода изменениям, что и астроциты, которые превращаются в элементы второго типа Альцгеймера.
В печени и извитых канальцах почек обнаруживаются изменения клеток, сходные с альцгеймеровскими. Наблюдаются также нервные клетки с хромолизом в различных стадиях «хронического заболевания» Ниссля (расплавление нервных клеток), реже поражение клеток, определяемых как «тяжёлое» и «отёчное». Воспалительные изменения не выражены. В основе клеточных изменений лежит нарушение клеточного обмена нуклеиновых кислот. Зёрна меди удается обнаружить в различных отделах мозга, больше всего в подкорковых узлах, в наиболее грубо измененных клетках внутри и межклеточно.
Первично страдает печень, изменения в мозгу обусловлены поражением печени. Клинически заболевание печени предшествует неврологическим симптомам.
Течение
Все новорожденные имеют низкую концентрацию церулоплазмина в крови и высокую концентрацию меди в печени. Однако если у здоровых детей концентрация церулоплазмина повышается, а концентрация меди снижается и к концу первого года жизни эти показатели становятся такими же, как у взрослых, то при болезни Вильсона этого не происходит.
До 6 лет болезнь, как правило, протекает бессимптомно. Первые симптомы обычно обнаруживают в подростковом возрасте, и в отсутствие лечения клиническая картина со временем развивается у всех больных.
Почти в 50% случаев болезнь начинается с одного из четырех вариантов поражения печени. Острый гепатит обычно проходит без лечения, и его нередко принимают за проявление вирусного гепатита или инфекционного мононуклеоза. Позже эту стадию можно легко упустить при сборе анамнеза.
Хронический гепатит развивается вслед за острым или независимо от него, клинически и гистологически он неотличим от хронического активного гепатита и приводит к циррозу печени. Иногда цирроз печени является первым проявлением заболевания без каких-либо признаков заболевания печени в анамнезе. Для острого некроза печени характерны быстро нарастающая желтуха, асцит, энцефалопатия, гипоальбуминемия, гипопротромбинемия, умеренное повышение активности печеночных ферментов в плазме и гемолитическая анемия с отрицательной пробой Кумбса. Острый некроз печени, как правило, заканчивается смертельным исходом.
В большинстве остальных случаев болезнь начинается с неврологических или психических нарушений. У больных всегда обнаруживают золотистые или зеленовато-золотистые кольца Кайзера—Флейшера — отложения меди в задней пограничной пластинке роговицы.
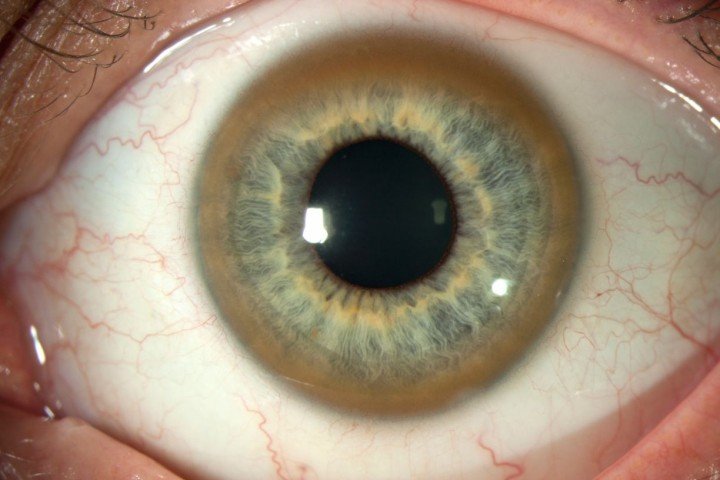
зеленовато-золотистые кольца Кайзера—Флейшера
Кольца Кайзера—Флейшера не влияют на зрение, но свидетельствуют о высвобождении меди из печени и возможном поражении мозга. Если опытный окулист с помощью щелевой лампы не может выявить колец Кайзера—Флейшера у больного с выраженными неврологическими и психическими нарушениями, то болезнь Вильсона можно исключить. Изредка кроме колец Кайзера—Флейшера обнаруживают халькоз хрусталика.
Неврологические нарушения при болезни Вильсона включают интеуционный тремор, тремор покоя, спастичность, мышечную ригидность, хорею, слюнотечение, дисфагию и дизартрию. Иногда наблюдаются патологические разгибательные рефлексы, а брюшные рефлексы нередко отсутствуют. Несмотря на то что медь откладывается во всех структурах мозга, нарушений чувствительности при болезни Вильсона никогда не бывает. Больные могут жаловаться на головную боль.
У большинства больных наряду с неврологическими наблюдаются и психические нарушения. Они могут проявляться бредовыми психозами, манией, депрессией или неврозами, но чаще всего странности в поведении больного не удается классифицировать. Психические нарушения обычно исчезают при устранении избытка меди, но иногда приходится проводить психотерапию или назначать психотропные средства.
Лишь в 5% случаев начало болезни бывает иным, то есть поражения печени и ЦНС отсутствуют. У женщин болезнь может начаться с первичной или вторичной аменореи или повторных необъяснимых самопроизвольных абортов, возможно, связанных с высоким содержанием свободной меди в эндометрии. Иногда кольца Кайзера—Флейшера обнаруживают случайно при визите к окулисту.
Какие изменения мозга вызывают признаки болезни?
Гепатоцеребральная дистрофия вызывает в тканях головного мозга размягчение нескольких ядерных образований (лентикулярного, хвостатого, зубчатого, подбугорных), мозжечка, глубинных слоев коркового слоя. В них образуются мелкие кисты.
Характерные токсические изменения различают:
- сосудистые — атония мелких сосудов способствует застойным явлениям крови, отеку окружающей ткани, гибели клеток, кровоизлияниям с выделением гемосидерина;
- клеточные — дистрофии подвергается макроглия нейронов, возможно формирование глии Альцгеймера, со временем клетки гибнут.
Установлено, что клеточные нарушения более распространены при позднем начале болезни и медленном течении. В печени атрофический процесс приводит к гибели гепатоцитов, целых долек с образованием некротизированных узлов. Между ними остаются очаги восстановления, нормальные клетки.
 Образуются новые сосуды, которые формируют связи между ветками воротной вены печени и руслом нижней полой
Образуются новые сосуды, которые формируют связи между ветками воротной вены печени и руслом нижней полой
Токсическое действие меди при болезни Вильсона
Острое отравление. Попадание внутрь солей меди в количестве, выраженном граммами, вызывает тяжелое поражение ЖКТ и других органов; иногда развивается некроз печени. Однако чаще всего рвота и понос, вызванные попаданием в ЖКТ солей меди, защищают организм больного от тяжелых последствий отравления.
Хроническое поражение. Избыток меди в печени может быть следствием не только болезни Вильсона; он возможен при первичном билиарном циррозе, атрезии внепеченочных желчных путей, индийском детском циррозе и других заболеваниях, сопровождающихся хроническим холестазом. Избыток меди в печени может обострять течение основного патологического процесса за счет прямого повреждения органелл гепатоцитов или стимуляции фиброза.
Общая картина заболевания
Второе название данного заболевания – гепатоцеребральная дистрофия или «медный токсикоз». Впервые это заболевание было изучено в 1912 году британским ученым-медиком Вильсоном. Несколько позже его труды были переработаны и дополнены отечественным медиком Коноваловым. Отсюда и название болезни Вильсона-Коновалова.
Болезнь Вильсона в основном «атакует» молодых мужчин до 25 лет и подростков. Чаще всего заболевание проявляет симптоматику в возрасте 10-15 лет.
Болезнь Вильсона-Коновалова имеет наследственный характер, то есть если у кого-то из родителей имело место быть такое заболевание, вероятность, что дети могут им заболеть, увеличивается в несколько раз.
Виновником данной болезни становится «сбой» в генотипе – один из генов, размещенных в 13-ой хромосоме, является дефектным. Именно он отвечает за нормальный обмен меди в организме. Если он «работает некорректно», в теле человека начинает вырабатываться и накапливаться избыточное количество металла (меди), которое постепенно отравляет весь организм. Медь откладывается в головном мозге и печеночных тканях.
Болезнь передается только от родителей к детям, но необязательно у кого-то из родителей будет присутствовать такое заболевание. Они могут быть носителями дефектного гена и передать его ребенку, у которого и проявится Вильсон-Коновалов. Когда один или оба родителя являются носителями, болезнь получит развитие только у ¼ младенцев.
Инфекционный характер заболевания полностью отсутствует, то есть контактирующие с больным люди не подвержены опасности.
Отмеченные случаи заболевания достаточное редкие – не более 3 больных на 100 000 здоровых человек.
Клиническая картина Вильсона-Коновалова такова, что:
- В организме начинается «сбой» процессов обмена меди, из-за чего начинается беспорядочная выработка меди, не выводящейся своевременно. Наблюдается избыток данного элемента;
- Повреждению сильнее всего подвержены: печень, головной мозг, органы зрения и почки;
- Снижается количество минеральных веществ в организме, что приводит к размягчению костных тканей и переломам.

Отсутствие лечения и своевременной диагностики приводит к летальному исходу. Обычно человек доживает до 30 лет. Смерть наступает в случае развития печеночной и почечной недостаточности.
Лечение Болезни Вильсона-Коновалова (гепатоцеребральной дистрофии):
- Диета № 5 — с ограничением меди до 1 мг в сутки — исключение шоколада, орехов, сухофруктов, раков, печени, цельной пшеницы.
- Препаратом выбора является купренил (пеницилламин), который эффективен в 90 % случаев. Д-пеницилламин или унитиол.
- Унитиол
- Витамин В6
Патогенетическое лечение при гепатолентикулярной дегенерации направлено на увеличение выведения меди из организма. Для этого применяются комплексоны (тиоловые соединения). Наиболее эффективным оказался пеницилламин. Его следует принимать постоянно по 1,5-2 г внутрь ежедневно.
Лечение пеницилламином сопровождается заметным улучшением состояния больных или даже приводит к полной ликвидации симптомов. Вполне удовлетворительные результаты получены и при применении унитиола.
Терапия патологии
Болезнь Вильсона сопровождается увеличением количества меди в кровотоке. Лечение разрабатывается таким образом, чтобы снизить ее концентрацию. Пациентам показано придерживаться диетического питания, которое заключается в исключении богатых медью продуктов. Пациентам не рекомендуется употребление изделий из какао и шоколада. Они должны отказаться от грибов и печени. Употребление любых видов орехов специалисты запрещают.
В период протекания болезни рекомендовано применение медикаментозной терапии. Наиболее часто больным делают назначение Пеницилламина. Суточная дозировка препарата составляет 1,2-2 грамма. Она определяется доктором в соответствии с индивидуальными особенностями пациента и степенью тяжести протекания патологии. При регулярном применении лекарства можно добиться стойкого клинического улучшения и даже полного устранения симптоматики. Параллельно рекомендовано принимать препараты, в состав которых входит большое количество витамина В6.
Лечение Пеницилламином требует придерживаться специальной схемы. На начальных этапах больному назначают 150 миллиграмм препарата через день. Такой схемы пациент должен придерживаться 7 дней. Следующую неделю лекарство принимается в такой же дозировке ежедневно. Далее рекомендовано каждую неделю повышать суточную дозировку медикамента на 150 миллиграмм. Максимальная доза препарата составляет 2 грамма. После улучшения состояния больного рекомендуется придерживаться поддерживающей дозировки 450-600 миллиграмм в сутки. Витамин В6 принимается в дозе 25-50 миллиграмм.
При возникновении нежелательных эффектов в виде тошноты, аллергии, болезней почек не рекомендован прием Пеницилламина. После этого прием препарата проводится в минимальной дозировке. Одновременно рекомендуется принимать 20 миллиграмм Преднизолона ежедневно. Длительность терапии заболевания составляет 10 суток. Если организм человека не принимает Пеницилламин, то рекомендовано назначение цинка Сульфата. Его необходимо принимать три раза в день по 200 миллиграмм.
Если у пациента диагностируется непереносимость Пеницилламина, то ему показан прием Унитола. Прием лекарство должен проводиться в течение месяца. После этого делают перерыв на 3 месяца. При использовании лекарства состояние человека улучшается, а симптоматика болезни стихает. Если в период протекания патологического процесса доминируют гиперкинезы, то больному рекомендовано принимать нейролептики короткими курсами. При возникновении ригидности пациентам показана:
- Леводопа;
- Тригексифенидил;
- Карбидопа.
Если консервативное лечение патологии не приносит желаемых результатов, а заболевание у пациентов протекает в тяжелой форме, то рекомендовано проведение хирургического вмешательства. В современных зарубежных клиниках проводятся операции по пересадке печени. Если исход хирургии положителен, то это приводит к улучшению состояния пациента и восстановлению процесса обмена меди в организме. В дальнейшем для терапии патологического процесса рекомендовано применение иммуносупрессивной терапии. В клинической практике нашей страны все чаще применяется биогенмоперфузия. При этом методе используются изолированные живые клетки печени и селезенки. При условии правильного проведения процедуры будет гарантирована ее высокая эффективность.
Схема лечения при патологии должна назначаться только доктором, что положительно отобразится на результате.
Определение болезни. Причины заболевания
Болезнь Вильсона-Коновалова — редкое наследственное дистрофическое заболевание, которое развивается из-за избыточного накопления меди в организме. Больше всего металла скапливается в головном мозге, глазах (с образованием характерной пигментации), печени и почках. При этом в органах нарушается обмен веществ и прогрессируют дистрофические изменения .
Заболевание впервые описано в 1912 году английским неврологом Семюэлем Вильсоном. В своём печатном труде он описал симптомы и особенности изменения внутренних органов при этом заболевании. Обычно первые проявления становились заметными в молодом возрасте, позже прогрессировала ригидность, сложности при глотании, снижение двигательной способности мягкого нёба, языка, губ (причина расстройства речи — дизартрии). Развиваются непроизвольные движения, расстройства психики — пониженное или необоснованно повышенное настроение, немотивированная агрессия, которая на поздних стадиях сменяется безучастностью, бредовые идеи, галлюцинации.
Российский невролог Николай Васильевич Коновалов изучал болезнь Вильсона на протяжении многих лет. Это позволило ему создать оригинальную полноценную классификацию различных форм заболевания. За огромный вклад Коновалова в исследование проблемы фамилия учёного навсегда дополнила название болезни.
Частота встречаемости — от 1 до 9 случаев на 100000 населения.
Основная причина возникновения болезни — мутация гена с названием ATP7B, который ответственен за встраивание ионов меди в белок церулоплазмин . Всего описано более 300 мутаций этого гена. База данных мутаций постоянно расширяется новыми вариантами . По международным оценкам, носителем аномального гена является примерно 1 человек из 100. При данном виде наследования заболевание проявляется симптоматикой только в том случае, если патологический ген был унаследован от обоих родителей. Мальчики и девочки болеют с одинаковой частотой .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Симптомы болезни Вильсона
Симптомы болезни Вильсона многообразны. Состояние присутствует при рождении, но симптомы не проявятся, пока не накопиться слишком много меди в печени, головном мозге или других органах.
Некоторые пациенты не испытывают признаков и симптомов гепатоцеребральной дистрофии до постановления диагноза и лечения. Однако, если симптомы присутствуют, они связаны с печенью, нервной системой и психическим здоровьем, глазами или другими органами.
Печеночные симптомы
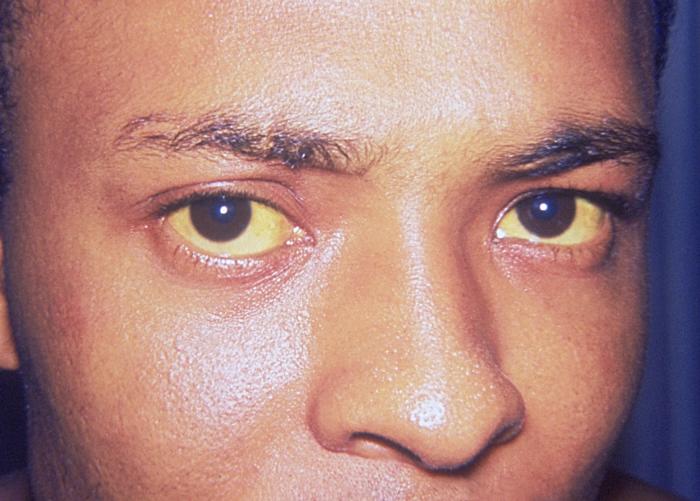
У людей с болезнью Вильсона могут развиться признаки гепатита или воспаление печени. В некоторых случаях эти симптомы у больных появляются, когда возникает острая печеночная недостаточность.Симптомы включают:
- чувство усталости;
- тошнота и рвота;
- отсутствие аппетита;
- боль над печенью, в верхней части живота;
- потемнение цвета мочи;
- светловатый ;
- желтоватый оттенок склер глаз и кожи (желтуха)
Некоторые люди с заболеванием имеют симптомы только в том случае, если у них развивается хроническая болезнь печени и осложнения, вызванные циррозом печени. Эти симптомы могут включать:
- чувство усталости или слабости;
- беспричинное похудение;
- вздутие живота от скопления жидкости в животе (асцит);
- отек голеней, лодыжек или ступней;
- зуд кожи;
- желтуху.
Признаки со стороны нервной системы и психического здоровья
У пациентов с заболеванием могут развиться проблемы с нервной системой и психическим здоровьем. Эти проблемы чаще встречаются у взрослых, но иногда появляются и у детей. Симптомы нервной системы могут включать:
- проблемы с речью, глотанием или физической координацией;
- скованность мышц;
- тремор или неконтролируемые движения.
Симптомы психического здоровья могут включать:
- тревогу;
- изменения в настроении, личности или поведении;
- депрессию;
- психоз.
Глазные симптомы
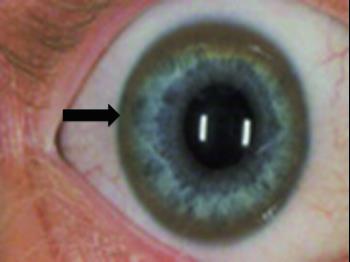
Многие люди с болезнью Вильсона имеют кольца Кайзера-Флейшера, представляющие собой , золотого или коричневатого цвета по краям роговицы. Кольца появляется в результате накопление меди в глазах. Так просто их не заметить, обычно врач-офтальмолог обнаруживает кольца во время специального обследования глаз, называемого осмотром щелевой лампой.
Другие симптомы и проблемы со здоровьем
Гепатоцеребральная дистрофия может повлиять и на другие части тела, вызывая симптомы или проблемы со здоровьем, в том числе:
- тип анемии, называемой гемолитическая анемия;
- проблемы с костями и суставами, например, артрит или остеопороз;
- проблемы с сердцем, например, кардиомиопатия;
- проблемы с почками (ацидоз почечных канальцев и нефролитиаз).
Лечение
Весь процесс терапии проходит под контролем врача-терапевта, который при необходимости подключает других специалистов, таких как, офтальмолог, невролог, кардиолог или гастроэнтеролог. Все зависит от того, какой орган пострадал от БВК. Необходима также обязательная консультация генетика.
С момента постановки диагноза лечение продолжается всю жизнь, так как это наследственное заболевание. Главным медикаментом, который назначается при «медном бунте» в организме является пеницилламин (купренил). Этот препарат принимается сначала по одной капсуле в сутки только после еды, постепенно дозу увеличивают до 6-10 капсул в течение трех месяцев. Принимать купренил нужно всю жизнь, с перерывом на 2-3 недели, если появляется побочное действие лекарства.
Так как медь перекрывает поступление в организм витаминов группы B, то параллельно назначаются витамины В1 и В6.
Лечение проводится под строгим контролем врача. Раз в неделю сдается общий анализ крови, биохимический — раз в 4 недели.
Весомой частью лечения является пожизненное соблюдение диеты №5. В ней исключены из меню продукты богатые медью. Подробнее о диете мы расскажем в следующем разделе. Также необходимо полностью отказаться от алкоголя.
Лечение
Схема лечения Вильсона-Коновалова подбирается в индивидуальном порядке. Основная задача медикаментозной терапии заключается в выведении избыточного количества меди из организма пациента.
Программа лечения от болезни Вильсона-Коновалова состоит из следующих этапов:
- Диагностика со сбором требуемых анализов и тестов;
- Получение консультации у специалистов с назначением индивидуальной схемы лечения;
- Осуществление медикаментозных и лечебных процедур для устранения симптомов заболевания;
- Прохождение реабилитационных мер в отделении неврологии.
Основные методы лечения Вильсона-Коновалова:
- Прием Д-пенициламина, уменьшающего количество меди в кровяных тельцах больного. Данный вариант лечения относится к патогенетическому;
- Реабилитационные меры в отделении неврологии для устранения негативного воздействия на нервную систему больного;
- Внутренний прием сорбентов и гепатопротекторов (при наличии повреждений печеночных тканей);
- Трансплантация печени при наличии серьезных, не возобновляемых повреждений органа;
- С помощью специализированного оборудования осуществляются процедуры по очистке плазмы крови от токсинов и свободной меди (гемоиммуносорбция), удалению из плазмы продуктов патологического распада (мембранный плазмаферез). Пациенту могут быть назначены до 3 процедур п очистке крови. Данная методика лечения зарекомендовала себя, как максимально эффективная.
Первые результаты от очистки крови заметны уже после 14 календарных дней. Медикаментозное лечение может длиться до полугода.
Кроме того, любая схема лечения от болезни Вильсона-Коновалова сопровождается приемом медикаментов. Среди них чаще всего выписывают:
Лекарственные препараты, снижающие количество меди в организме больного (хелаты);
Медикаменты, способствующие регенерации печеночных клеток;
Желчегонные лекарственные медикаменты;
Комплекс витаминов (важно применение В6);
Лекарства, блокирующие поступление меди в организм с продуктами питания (например, содержащие цинк);
Медикаменты с антиоксидантными свойствами, снижающие интоксикацию организма;
Средства, подавляющие действие иммунной системы;
Лекарственные препараты успокоительного характера для устранения неврологических расстройств;
Средства, обладающие противовоспалительным результатом.
Назначенные доктором медикаменты пациент обязан принимать с начала диагностирования болезни и до конца его жизни. Если больной снизит дозировку или полностью прекратит лечение, то Вильсон-Коновалов может вернуться с более сильной симптоматикой.
В запущенных случаях заболевания, когда медикаментозные методы не дают должного результата, а состояние больного ухудшается диагностирована печеночная недостаточность, назначается трансплантация печени. Новая печень приживается и нормально функционирует уже через месяц после операции.
Обратите внимание
Народная медицина бессильна в борьбе с синдромом Вильсона. Если вам предлагают «эффективное» народное средство, не верьте! Потеряв время на бесполезное самолечение, можно подвергнуть риску жизнь больного! Это нарушение успешно лечится лишь современными медицинскими препаратами
Важно, чтобы схему подбирал опытный врач
Нужна строгая диета (стол 5а). Ее цель – сохранить функции печени. Полностью нужно исключить продукты, в которых содержится много меди (бобовые, орехи, кофе, шоколад и т.д.).
Основное лечение заключается в регулярном приеме препаратов, которые способны выводить медь. Это, в первую очередь, D-пеницилламин. Отечественный его аналог не подходит. Он высокотоксичен. Так как принимать препарат нужно всю жизнь, со временем появляются побочные эффекты (анемия, дерматиты и др.). Сейчас предлагается альтернативное лечение при помощи солей цинка. Можно также комбинировать препараты.
Такие препараты назначают по особой схеме. Доза постепенно увеличивается. Лечение будет пожизненным.
За рубежом для борьбы с этой патологией начали практиковать пересадку печени. Она используется при тяжелом течении заболевания, если консервативные методы оказались бессильны. После нее уже не нужно будет принимать препараты.
Также крайне важно гепатопротекторное лечение. Его задача – максимально сохранить функции печени
Чтобы сохранить функции мозга, потребуется наблюдение у невролога.
Если правильно подобрать методы лечения, возможно полное выздоровление (80% всех случаев) или заметное улучшение. Но столь положительный эффект наблюдается лишь в том случае, если заболевание выявлено на самой ранней стадии. При этом еще не произошло катастрофичного отравления органов и тканей токсичной медью, не появились мучительные неврологические симптомы.
Выводы
Восстановительное лечение пациентов с неврологической стадией БВК необходимо проводить в течение всей жизни с учетом формы и стадии болезни, а также клинической картины и данных лабораторных исследований.
Хелаторы пеницилламин и триентин являются препаратами выбора в начале лечения неврологической стадии БВК, при этом начинать применение пеницилламина следует с низких доз и под контролем выделения меди с мочой. На этапе поддерживающей терапии лучше применять комбинированное лечение пеницилламином в невысокой дозе и солями цинка (цинка сульфат). При лечении пеницилламином необходимо дополнять терапию витаминами группы В.
Соли цинка можно считать препаратом выбора при пресимптомной стадии болезни, а также на этапе поддерживающей терапии как монопрепарат или в комбинации с пеницилламином. С учетом того что лечение БВК пожизненное, весьма важным является доступная стоимость препаратов цинка.
Трансплантация печени при БВК показана лишь пациентам с острой или прогрессирующей печеночной недостаточностью, которая не поддается консервативному лечению.
На протяжении всей жизни из рациона лиц с БВК следует исключить продукты с высоким содержанием меди.
В зависимости от клинической картины и данных лабораторных исследований, желательно 1–2 раза в год проводить курсовую терапию с применением нейропротекторов, гепатопротекторов, энтеросорбентов, антигипоксантов, метаболиков, вазоактивных и антиоксидантных препаратов, а также лечебно-реабилитационные мероприятия.