«кукольные дети, которые всегда улыбаются»
Содержание:
- Лечение болезни Ангельмана
- Симптомы и признаки
- Симптомы и признаки
- Уход за ребенком и прогноз заболевания
- Причины и провоцирующие факторы
- Риски
- Причины
- 2 Клинические проявления
- Примечания
- Генетические мутации повышают риск появления заболевания
- Формы
- Синдром Ангельмана у детей – симптомы
- Диагностика
- Прогноз
- Лечение
- Симптомы синдром Ретта
Лечение болезни Ангельмана
На сегодняшний день терапия синдрома Ангельмана направлена на устранение/смягчение симптомов и имеет поддерживающий характер. Специфических препаратов или методов генетической коррекции, способных излечить заболевание, не имеется. В большинстве случаев пациенты с этим синдромом обладают относительно хорошим общим физическим здоровьем, а детям вполне возможно оказывать стандартный педиатрический уход, включая иммунизацию.
В случаях развития приступов, рекомендован прием противосудорожных средств. Как правило, такие проявления заболевания удается адекватно контролировать при помощи одного препарата. Впрочем, иногда купирование подобной симптоматики требует сочетания нескольких медикаментов.
Поэтому необходимо консультирование с лечащим врачом, который назначит наиболее действенное средство/набор средств, исходя из индивидуальных особенностей пациента.
При нарушениях сна рекомендовано воспользоваться методами поведенческой терапии и строго соблюдать процедуры, позволяющие нормализовать отдых. В течение определенного периода может оказаться полезным прием седативных препаратов.
Устранение проблем с кормлением возможно с применением различных методов и средств. Например, затруднения при грудном вскармливании помогут откорректировать специальные соски для детей с угнетенной способностью к сосанию.
Устранить гастроэзофагеальный рефлюкс способны препараты, стимулирующие продвижение пищи по пищеварительному тракту (мотилитики). В отдельных случаях рекомендована хирургическая коррекция сфинктера пищевода – клапана, который соединяет пищевод с желудком.
Фиксаторы и ортезы для голеностопного соединения эффективны при освоении навыков ходьбы. Примерно в 10% случаев синдрома Ангельмана наблюдается приобретенный сколиоз. Для его устранения возможно применение брекетов либо организация хирургического вмешательства. Кроме того, хирургическим путем обычно производится лечение косоглазия.
Коррекция психо-эмоционального состояния пациентов и социализация.
Одними из ключевых факторов в терапии синдрома Ангельмана являются приемы своевременной адекватной
- Физиотерапии
- Эрготерапии
- Коммуникационной терапии
- Поведенческой терапии
Большинство людей, подверженных синдрому счастливой куклы, на протяжении жизни будут испытывать последствия значительных задержек в развитии, у них сохранится ограниченная речь и затрудненная моторика.
Однако методы, которые применяют перечисленные варианты терапий, позволяют таким людям достичь максимального потенциала в своем развитии. И чем раньше подключить указанные методики, тем благоприятнее прогноз.
По мере старения у пациентов с болезнью Ангельмана не наблюдается поведенческая регрессия и редуцирование приобретенных навыков.
Отдельно следует отметить важность развития коммуникативных/речевых способностей. Выше отмечен крайне скудный набор слов, которыми пользуются «Петрушки»
Иногда речь у них вовсе не развита. Но особенность этих пациентов в том, что, как правило, возможности их понимания/интерпретации получаемых сведений заметно превышают освоенный набор средств выражения.
Потому эффективным способом социализации и повышения уровня коммуникации для них является освоение альтернативных способов общения – например – языка жестов.
Симптомы и признаки
Клиническая картина недуга весьма разнообразна и может отличаться в каждом конкретном случае. В частности, у ребенка проявляются физические и психические отклонения, некоторые признаки характерны для всех случаев, другие встречаются значительно реже.
|
Физические отклонения |
Психические отклонения |
|
|
Клинические проявления недуга принято делить также в зависимости от их распространенности. Так, существуют симптомы, которые встречаются у всех детей, страдающих данным недугом, признаки, возникающие в большинстве случаев (но не всегда) и более редкие проявления.
|
Характерные симптомы |
Частые признаки |
Более редкие проявления |
|
|
|
Клинические проявления недуга меняются по мере взросления ребенка. Так, у детей подросткового возраста наблюдается задержка полового созревания (при этом детородная функция в дальнейшем не нарушается), а когда ребенок становится взрослым, он выглядит значительно моложе своих сверстников.
Нередко у детей старшего возраста возникают и другие проявления заболевания, такие как нарушение мочеиспускания, сложные нарушения моторики рук, проблемы с общением (подросток понимает устную речь, но не видит необходимости отвечать собеседнику).
Какова продолжительность жизни детей с синдромом Вильямса? Ответ узнайте прямо сейчас.
Симптомы и признаки
Клиническая картина недуга весьма разнообразна и может отличаться в каждом конкретном случае. В частности, у ребенка проявляются физические и психические отклонения, некоторые признаки характерны для всех случаев, другие встречаются значительно реже.
| Физические отклонения | Психические отклонения |
|
|
Клинические проявления недуга принято делить также в зависимости от их распространенности. Так, существуют симптомы, которые встречаются у всех детей, страдающих данным недугом, признаки, возникающие в большинстве случаев (но не всегда) и более редкие проявления.
| Характерные симптомы | Частые признаки | Более редкие проявления |
|
|
|
Клинические проявления недуга меняются по мере взросления ребенка. Так, у детей подросткового возраста наблюдается задержка полового созревания (при этом детородная функция в дальнейшем не нарушается), а когда ребенок становится взрослым, он выглядит значительно моложе своих сверстников.
Нередко у детей старшего возраста возникают и другие проявления заболевания, такие как нарушение мочеиспускания, сложные нарушения моторики рук, проблемы с общением (подросток понимает устную речь, но не видит необходимости отвечать собеседнику).
Уход за ребенком и прогноз заболевания
Болезнь обнаруживают в основном, когда ребенок находится в возрасте от трех до семи лет. Симптоматика выражена четко именно к достижению этого возраста, не раньше. Поскольку симптомы неспецифичны, тяжело диагностировать рассматриваемый синдром у младенца
Важно отметить, что болезнь относительно редкая. Потому врач, есть вероятность, не сможет и догадаться, что он имеет дело с синдромом Ангельмана
В Европе есть больше данных про рассматриваемую патологию, потому диагностика у них более качественная и быстрая. Поставить диагноз должен врач-генетик, который назначает генетический анализ, на основе которого делаются выводы.
Прогноз болезни коррелирует со степенью поражения пятнадцатой хромосомы. Часть пациентов нормально ведут себя в быту, общение у них также почти нормальное. Но в части случаев ребенок не может ходить, речь у него ответствует. Люди с синдромом Ангельмана живут 20-50 лет. Качество жизни во многом зависит от тяжести синдрома, как уже было сказано выше.
Причины и провоцирующие факторы
Основной причиной развития недуга считается изменение или отсутствие одного из фрагментов 15 хромосомы. Риск развития патологии у ребенка повышается, если у его близких родственников (родителей) имеются следующие генетические отклонения:
- Присутствие лишних хромосом (одной или нескольких).
- Хромосомная инверсия, когда один из участков хромосомы перевернут, при этом часть его фрагмента утеряна, а гены располагаются в противоположном порядке.
- Перестройка Y-хромосомы, когда 2 ее участка меняются местами, при этом некоторые фрагменты мутировавшей хромосомы могут отсутствовать.
- Утрата одного или нескольких хромосомных фрагментов.
- Присоединение фрагмента одной хромосомы к другой.
- Наличие лишних генов, возникших в результате репликации одной или нескольких хромосом.
- Кольцевое строение хромосомы, когда концы ее соединяются между собой, образуя кольцо.
Риски
Оценка риска повторного рождения ребенка с синдромом Ангельмана у тех же родителей очень сложна, необходима консультация профессионального генетика. Считается, что обычная делеция является спонтанной, риск повтора меньше 1 %. В случае молекулярной микроделеции в 15q11-q13, если она наблюдается и у матери, риск теоретически до 50 %. Мутации внутри гена UBE3A могут быть случайными и неунаследованными, в этой ситуации риск повтора <1 %; однако эти мутации можно унаследовать от нормальной матери, и тогда теоретический риск 50 %. Партеногенетическая дисомия 15-й пары — случайная ситуация; риск повтора <1 %. Есть несколько людей с AS с необычными преобразованиями хромосомы 15, включая область 15q11-q13; в этих случаях оценка риска повтора зависит от хромосомных нарушений у родителей.
Причины
При данном заболевании отсутствуют отдельные гены 15-й хромосомы. Большинство случаев характеризуется частичной делецией или же мутацией 15-й хромосомы. При синдроме Ангельмана страдает материнская хромосома, а в случае изменений в отцовской хромосоме развивается синдром Прадера — Вилли. Зачастую синдром провоцируется спонтанным хромосомным дефектом, который характеризуется отсутствием смежной области, состоящей из четырех миллионов пар оснований ДНК в местах q11- q13 15-й хромосомы. Синдром Ангельмана имеет кариотип 46 XX или XY, 15р. Независимые исследования к причинам синдрома Ангельмана относят мутации в гене UBE3A. Ферментный компонент сложной системы деградации белков считают продуктом этого гена.

2 Клинические проявления
Признаки синдрома проявляются в период 6-12 месяцев жизни. Его трудно распознать до этого срока, потому что болезнь сопровождается задержкой развития организма: как физического, так и умственного. Явные симптомы AS появляются только по достижении ребенком возраста 2 лет.
Как правило, пациенты с AS имеют счастливый вид. Они любят играть в различные игры и общаться, хотя сначала им может быть несколько трудно установить связь с другими людьми. Поскольку дети с AS постепенно развиваются (у них появляются различные способности и определенный характер), окружающие их начинают лучше понимать.

Специалистами были разработаны критерии диагностики AS. Они представлены в таблице ниже.
Симптомы, полезные в качестве вспомогательных критериев (их отсутствие не исключает диагноз AS)
Обязательные признаки (встречаются у 100% пациентов с AS)
Часто встречающиеся признаки (более чем у 80% пациентов с AS)
Дополнительные признаки (частота встречаемости 20 -80%)
Нормальное течение перинатального периода, окружность головы при рождении соответствует гестации, выраженные пороки развития отсутствуют
Выраженная задержка психического развития
Задержка прироста окружности головы, в результате чего формируется абсолютная или относительная микроцефалия (маленький череп) к двухлетнему возрасту
Плоский затылок с «канавкой». Прогнатизм (вперед выступающая нижняя челюсть). Косоглазие. Широкий рот с редкими широкими зубами. Сколиоз.
Очевидная задержка развития, начиная с 6-12-месячного возраста
Речевые нарушения (минимальное использование слов или отсутствие речи )
Эпилепсия. Появление судорожного синдрома обычно с трехлетнего возраста. Уменьшается тяжесть судорожного синдрома с возрастом, но может проявляться на протяжении всей жизни
Высунутый язык, нарушения сосания, особенно в период грудного вскармливания
Отсутствие прогрессирующей потери приобретенных навыков
Невербальные коммуникативные навыки развиты лучше, чем вербальные
ЭЭГ-паттерн в виде высоко амплитудных разрядов медленных комплексов, острая-медленная волна частотой 2-3 Гц, провоцируемая нередко закрыванием глаз
Чрезмерное жевание. Частое слюнотечение. Запор. Ожирение (в старшем возрасте)
Нормальные результаты гематологических, метаболических и биохимических лабораторных тестов
Атаксия или покачивания вперед, неустойчивость, неуклюжесть походки
Резкие, быстрые движения
—
Гипопигментация кожи, светлые волосы и глаза (только в случаях делеции гена)
Отсутствие изменений структуры головного мозга по результатам КТ или МРТ (возможны проявления дисмиелинизации или умеренная кортикальная атрофия)
Тремор (дрожание) конечностей
—
Усиление сухожильных рефлексов с нижних конечностей
—
Специфические особенности поведения (частый немотивированный смех, быстрая возбудимость, стереотипии в виде размахивания руками, гиперактивность )
—
При ходьбе приподнятые плечи и полусогнутые в локтевых суставах руки
—
—
—
Сверхчувствительность к повышенной температуре окружающей среды
—
—
—
Повышенное внимание и «притяжение» к воде
—
—
—
Нарушение сна, снижение потребности во сне
Примечания
- X-сцепленная умственная отсталость. (рус.). Центр Молекулярной генетики при Медико-генетическом научном центре РАМН. Проверено 8 мая 2020.
- Yamasaki K., Joh K., Ohta T., Masuzaki H., Ishimaru T., Mukai T., Niikawa N., Ogawa M., Wagstaff J., Kishino T. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. (англ.) // Human molecular genetics. — 2003. — Vol. 12, no. 8. — P. 837—847. — DOI:10.1093/hmg/ddg106. — PMID 12668607.
- Petersen M. B., Brøndum-Nielsen K., Hansen L. K., Wulff K. Clinical, cytogenetic, and molecular diagnosis of Angelman syndrome: estimated prevalence rate in a Danish county. (англ.) // American journal of medical genetics. — 1995. — Vol. 60, no. 3. — P. 261—262. — DOI:10.1002/ajmg.1320600317. — PMID 7573182.
- Steffenburg S., Gillberg C. L., Steffenburg U., Kyllerman M. Autism in Angelman syndrome: a population-based study. (англ.) // Pediatric neurology. — 1996. — Vol. 14, no. 2. — P. 131—136. — DOI:10.1016/0887-8994(96)00011-2. — PMID 8703225.
Генетические мутации повышают риск появления заболевания
Синдром Ангельмана встречается нечасто — примерно 1 случай на 10 – 20 тыс. новорожденных малышей. Основной причиной патологии является потеря в 15 хромосоме копий нормальных материнских генов, что происходит по причине нарушения деления данной хромосомы. Кроме того заболевание может возникнуть в связи с мутацией отцовских генов, отцовской дисомией или трисомией.
В норме здоровый человек получает от матери и отца по одной копии 15 хромосомы. Если ребенок получил от кого-то из родителей генетически измененную копию (особенно от матери, так как у нее копии генов более сильные, чем у отца), у ребенка развивается синдром Ангельмана.
Заболевание носит имя Гарри Ангельмана, британского врача-педиатра, который впервые дифференцировал данный синдром в 1965. Тогда он получил название синдрома счастливой марионетки, однако сегодня этот термин не используется, так как это было признано пренебрежительным.
Доктор Ангельман занимался лечением нескольких детей со сходными симптомами и предполагал наличие общего диагноза. Доказать диагноз и получить точные данные на тот момент было не возможно, ввиду отсутствия технологий, которые доступны сегодня. Свои догадки врач отразил в статье под названием «Дети марионетки».
Появление синдрома Ангельмана связано с наличием у родителей будущего ребенка различных хромосомных аномалий. Среди таких отклонений обычно называют:
- трисомию хромосом – присутствие одной или нескольких лишних хромосом в хромосомном наборе;
- инверсию – разворот одного из участков хромосомы на 180 градусов, при этом часть хромосомы пропущена, а гены располагаются в противоположном порядке;
- микроделецию, которая является результатом перестройки Y-хромосомы и обмена участками между хромосомами, наблюдается небольшое количество хромосом, а также может отсутствовать один из генов;
- делецию – нехватку одного из участков хромосомы;
- транслокацию – перенос или присоединение участка одной хромосомы к другой хромосоме;
- дупликацию – копирование части хромосом, результатом чего становится лишний генетический материал;
- кольцевую хромосому – на концах хромосомы отсутствует генетический материал, при этом новообразованные концы соединяются в виде кольца.
Генные мутации, которые могут вызвать развитие синдрома
Рассматриваемая болезнь обязана своим названием врачу-педиатру Гарри Ангельману, который впервые диагностировал отклонение в 1965 году и назвал пациентов кукольными детьми.
Именно картина «Мальчик-марионетка» подарила специалисту уверенность в собственной правоте. Картина отображает смеющегося мальчика, который очень напомнил Гарри его пациентов и он решает написать о них общую статью, которая в дальнейшем получила название «Дети-марионетки».
Признаки синдрома Ангельмана
В 1965 году издается научная работа, интерес о которой был забыт до восьмидесятых, когда упоминания о патологии появились в медицинской среде. Закономерность отсутствия части 15-й хромосомы была установлена только в 1987 году. Поскольку диагноз «синдром счастливой марионетки» унижал и пугал многих родителей, было решено использовать фамилию Ангельмана. Среди современных ученых, изучающих данное явление, стоит выделить М. Б. Миронова и К. Ю. Мухина.
В качестве факторов риска и провоцирующих элементов стоит рассмотреть родительские хромосомные аномалии.
- Кольцевая хромосома – удаление генетического материала с концов хромосомы и соединение новообразованных концов в кольцо.
- Дупликация — частичное повторение хромосом и соответствующее появление лишнего генетического материала.
- Транслокация – присоединение к хромосоме элемента другой хромосомы.
- Делеция предполагает отсутствие одного из секторов хромосомы.
- Микроделеция является следствием перестройки Y-хромосомы и обмена секторов между половыми хромосомами при мейозе. Немного хромосом при этом отсутствует, чаще всего одного из генов тоже нет.
- Инверсия – разворот на 180 градусов одного из участков хромосомы, порядок расположения генов обратный, а часть хромосомы опущена.
- Трисомии хромосом обусловлена наличием одной или больше лишних хромосом в наборе. Причиной такого генетического дефекта является нерасхождение хромосом при делении.
Формы
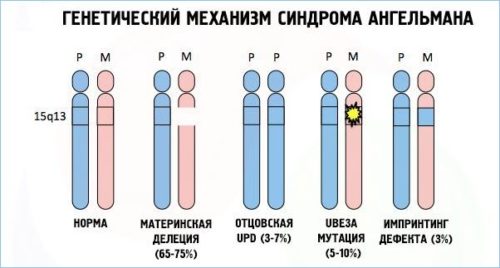
Синдром Ангельмана связан с четырьмя вариантами генетических мутаций:
- Вновь возникшей хромосомной мутацией, которая связана с потерей участка хромосомы в локусе 15 q11 — q13 . Данная мутация – причина около 80 % всех случаев заболевания.
- Одноотцовской дисомией, которая связана с потерей материнского локуса (отсутствие генетического материала матери). Данный вариант встречается редко (около 5% всех случаев).
- Дефектом ряда генов, подверженных геномному импринтингу (ГИ). Данные дефекты возникают у 2-4% больных в результате непосредственного нарушения импринтинга (различия в преобразовании информации гена в белок или РНК, которые зависят от происхождения гена). Чаще всего возникает в результате утраты центра регуляции ГИ. Дефекты ГИ без утраты центра регуляции являются результатом спонтанной мутации, повторение которой – большая редкость.
- Спонтанной мутацией материнской копии, которая вызывает отсутствие преобразования в мозге копии гена UBE3A. Данный ген кодирует деятельность убиквитинлигазы (фермент, участвующий в сложном процессе распада белков). Дефицит данного фермента относится к молекулярным механизмам синдрома.
Установить форму заболевания у 7-9 % в настоящее время не представляется возможным.
Синдром Ангельмана у детей – симптомы
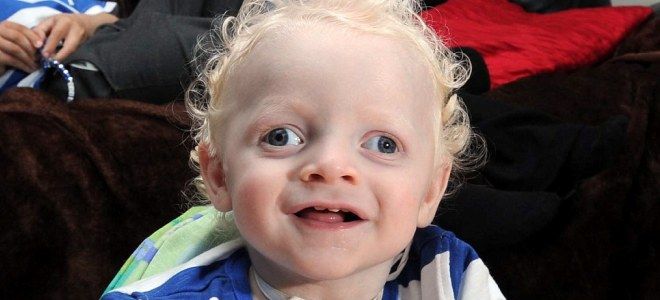
Первые признаки патологического нарушения начинают проявляться в возрасте 6 месяцев. По мере взросления малыша они становятся более отчетливыми. Синдром петрушки симптомы имеет такие:
- Группа физических признаков. У пациентов, которым поставлен данный диагноз, наблюдаются проблемы со зрением. Чаще это косоглазие, наличие пятен на радужке. К тому же у таких пациентов нарушена пропорциональность тела: небольшая голова по сравнению с другими частями туловища. В возрасте 12-ти месяцев начинает проявляться еще один сопутствующий признак – малыш широко открывает рот и слегка выпячивает язычок. У некоторых из них пациентов проявляется гипопигментация отдельных участков кожи.
- Неврологические признаки. У пациентов наблюдается расстройства сна. Одновременно отмечаются нарушения речевых функций. Малыша в возрасте 3 лет часто могут одолевать эпиприпадки. Однако тяжесть и интенсивность таких признаков снижается по мере взросления ребенка.
- Психологические признаки. У пациентов отмечается умственная отсталость и аффективное поведение.
Синдром Ангельмана – фенотипические признаки

Для этого заболевания характерна своя клиническая картина. Синдром петрушки болезнь сопровождается следующими симптомами:
- тяжелая задержка развития;
- тремор;
- нарушение равновесия;
- повышенная жажда;
- усиленные жевательные движения;
- плоские ладони;
- проблемы с мелкой моторикой (с трудом застегивают пуговицы, молнии);
- неконтролируемое мочеиспускание;
- лишний вес;
- заостренный подбородок;
- нижняя часть челюсти слегка выступает вперед;
- искривление позвоночника.
Диагностика
Диагностика заболевания основывается на сборе анамнеза (истории) заболевания, жизни больного, генетических изменений в гениалогическом древе, т.е наличия в семье/родственников данной патологии.
Основным методом исследования является генетический анализ, который указывает на дефектную X-хромосому и с помощью данного анализа подтверждается болезнь Ретта.
Проводят инструментальные методы исследования головного мозга:
- Компьютерная томография: с ее помощью анализируют структуры мозга и патологические нарушения. Выявляют экстрапирамидные расстройства (нарушение координации движений, нарушения речи и др.). А также имеются признаки отсутствия развития головного мозга.
- Электроэнцефалография исследуют структуры головного мозга, определяя биоэлектрическую активность. Замедленный фоновый ритм подтверждает наличие мутации Х-хромосомы.
При синдроме Ретта проводят дополнительные методы исследования, которые могут подтвердить или опровергнуть патологии других органов и систем на разных стадиях заболевания:
- Электрокардиография, эхокардиография свидетельствуют о патологии сердца. Могут быть аритмия, учащенный пульс и хроническая сердечная недостаточность по большому или малому кругам кровообращения.
- УЗИ органов брюшной полости свидетельствуют о патологии пищеварительного тракта, печени и желчевыводящих протоках. Реже выявляются патологии мочевыделительной системы.
- Электронейромиография: с ее помощью исследуют проведение импульсов через нервные окончания мышц, суставов и др.
Постановка диагноза на начальных этапах основывается на клинических проявлениях заболевания. Чем сильнее выражены клинические признаки в возрасте не соответствующей стадии, тем короче продолжительность жизни ребенка.
Дифференциальную диагностику синдрома Ретта у детей следует проводить с аутизмом. Что у синдрома Ретта что и у аутизма, есть одинаковые клинические проявления.
Аутизм – дегенеративное расстройство нервной системы, развивающееся при нарушении формирования головного мозга и проявляющееся дефицитом общения, ограничением интересов и стереотипно повторяющимися действиями.
| Признак | Синдром Ретта у детей | Аутизм |
| Нарушение физического развития с ярко выраженной клиникой, возраст – 1 год. | Практически не выявляется | Часто выражено |
| Стереотипность двигательных возможностей | Однообразные повторения движений рук, напоминающих мытье в пределах 1 пояса. | Разнообразные, многогранные движения рук с различными областями. |
| Одинаковые движения с предметами | Отсутствуют | Присутствуют. Например: щелканье ручкой, открытие/закрытие книги и др. |
| Нарушение координации (поражение мозжечка и ствола мозга) | Прогрессирующий характер, вплоть до неподвижности. | Движения напоминают манерные, но патологии мозжечка нет. |
| Эпилептиморфные припадки | Частые повторения в зависимости от стадии. | Редко или вообще отсутствуют. |
| Нарушение дыхания | Апноэ (отсутствие дыхания) в ночное или дневное время суток. | Отсутствуют. |
| Нарушение роста костей верхних и нижних конечностей, головы | Возникает. | Не возникает |
Прогноз
Прогноз при синдроме Ангельмана зависит от характера хромосомной аномалии и своевременности ее обнаружения. Тяжелее всего приходится тем деткам, чья 15 хромосома содержит «пропуски» генов (делеция). Вероятность ходить и разговаривать у таких пациентов чрезвычайно мала. Остальные случаи при внимательном подходе и любви к своему ребенку поддаются коррекции.
Стать полноценными членами общества такие больные, увы, не смогут, при всем том, что они далеко не глупы, понимают речь и ее смысл. Вот только проблемы с общением у них остаются на всю жизнь. Пациентов с детства можно обучить языку жестов, но нельзя заставить общаться при помощи слов. Лексикон «говорящих» больных ограничивается минимумом слов, употребляемых в быту (5-15 слов).
Что касается продолжительности жизни и общего состояния здоровья больных с синдромом Ангельмана, то здесь цифры колеблются на средних показателях. Во взрослой жизни пациенты в основном сталкиваются с такими проблемами со здоровьем, как сколиоз и ожирение, которые при правильном подходе к лечению не опасны для жизни.
Лечение
Синдром Ангельмана — неизлечимая генетическая аномалия. В настоящее время ученые-медики активно разрабатывают эффективные терапевтические методики. Если синдром был диагностирован во время внутриутробного развития, специалисты рекомендуют прервать беременность. Новорожденному больному ребенку необходим тщательный уход, забота и высококвалифицированная терапия.

Симптоматическое лечение помогает облегчить состояние больных с синдромом марионетки. Им назначают лекарственные препараты и немедикаментозные процедуры:
Антиконвульсанты снижают частоту и силу эпиприступов. Больным назначают одновременно несколько противосудорожных средств, поскольку приступам характерны несколько видов судорог. К наиболее популярным препаратам относятся: «Вальпроевая кислота», «Конвулекс», «Ламотриджин», «Клоназепам». Эти средства предупреждают судороги, улучшают настроение и психическое состояние больных.
Витаминотерапия — прием витаминов группы В, С, Д и Е. Лечение витаминами следует проводить только по назначению врача, поскольку они снижают эффективность противоэпилептических препаратов.
Снотворные препараты, улучшающие сон у легковозбудимых пациентов – «Мелатонин», «Дифенгидрамин».
При появлении проблем с пищеварением и стулом больным назначают слабительные средства – «Сенаде», «Слабилен», «Фитолакс», пре- и пробиотики – «Хилак форте», «Линекс», «Бифиформ».
Гормональная терапия показана в крайних случаях, когда невозможно другими способами скорректировать поведение больных
Им вводят гормон секретин, нормализующий пищеварение, положительно влияющий на внимание пациентов, а также окситоцин, улучшающий познавательные способности ребенка, память, поведение. Подобная схема лечения гормонами разработана американскими учеными для лечения детей, больных аутизмом.
При синдроме Ангельмана показана поведенческая терапия, работа с психологом, дефектологом и логопедом.
Физиотерапевтические процедуры помогают справиться с гипотонией мышц и проблемами с суставами
Обычно врачи назначают парафиновые аппликации, электрофорез, магнитотерапию. Увереннее стоять на ногах больным помогает ЛФК, профессиональный массаж, аквагимнастика в прохладной воде.
Чтобы избавиться от гиперсаливации, применяют препараты, угнетающие слюнообразование и оперативные вмешательства, направленные на реимплантацию слюнных протоков.
Средства народной медицины, как и гомеопатические препараты, слабоэффективны в лечении синдрома Ангельмана, но абсолютно безопасны. Сбор на основе пиона, солодки и ряски снижает частоту судорожных приступов, отвар лаванды и водный настой пустырника оказывают общее успокаивающее действие.
Симптомы синдром Ретта

После рождения ребенок ничем не отличатся от своих сверстников в течение первых месяцев своей жизни.
Врачи не могут заподозрить симптомы и выявить патологические изменения, Ретта.
Окружность головки соответствует норме.
Единственным признаком патологии может быть снижение и вялость мышечного тонуса. Также может изменяться температура тела (низкая, 35 градусов), бледные кожные покровы и высокая влажность ладоней.
К 5-6 месяцам начинает выявляться отставание в физическом развитии ребенка. Он не ползает, не может переворачиваться на спинку или на бок.
В большинстве случаев, в более старшем возрасте, ребенок не может удерживать свое тело в вертикальном положении и сидеть.
Патогомоничные (отличительные) признаки синдрома Ретта:
- специфические двигательные приемы рук (см. фото выше). Дети с данным синдромом не могут удерживать различные предметы в руках. У них выявляются одинаковые стереотипные движения, напоминающие перебор пальчиков или хлопки руками на уровне грудной клетки. Дети иногда ударяют себя ручками по различным частям тела.
- развитие умственной отсталости, отсутствует стремление изучать что-то новое. Дети до 1 года могут научиться ходить, говорить, узнавать окружающих (родителей), но в течение некоего временного промежутка теряют эти способности.
- уменьшение размеров головного мозга и окружности головы.
- развитие судорожных припадков, напоминающих эпилептические.
- искривление позвоночника из-за нарушения позвоночного сегмента, ведущего к мышечной дистонии;
- нарушение речи;
- замедленный рост;
- развитие патологий сердечно-сосудистой системы, пищеварительного тракта и др.;
- нарушение дыхания, ночью или днем во время сна могут наступать промежутки отсутствия дыхания;
- во время приема пищи с едой дети могут заглатывать воздух, провоцируя срыгивание.