Синдром вильямса (лицо эльфа)
Содержание:
- Синдром Ангельмана – диагностика
- Первые признаки синдрома Вильямса
- Формы синдрома Элерса-Данлоса
- Факторы-провокаторы синдрома Вильямса
- Борьба с гиперкальциемией
- Перспективы лечения синдрома Вильямса
- Причины синдрома Вильямса
- Патологии других органов и систем
- Чем болеют эльфы?
- Что делать?
- Приобретенный талант
- Симптомы и признаки
Синдром Ангельмана – диагностика
Обследованием детей с подозрением на синдром Ангельмана занимаются врачи-неврологи, психиатры и генетики. Родители предъявляют жалобы на отсутствие речи, двигательные стереотипии, трудности формирования ходьбы и других двигательных навыков, гиперактивность. Проводится дифференциальная диагностика, в ходе которой должны быть исключены более распространенные заболевания, такие как умственная отсталость, расстройства аутистического спектра, деменции, мутизм, несимптоматические формы эпилепсии. Комплексное исследование включает следующие процедуры:
- Общий осмотр. На наличие синдрома часто указывают специфические черты внешности пациентов: высунутый язык, слюнотечение, крупный рот, широкие редкие зубы, выступающая вперед нижняя челюсть, плоская форма затылка. Характерен светлый оттенок кожи, глаз и волос. Походка детей напоминает движения куклы-марионетки из-за усиления сухожильных рефлексов и снижения мышечного тонуса.
- Осмотр психиатром. В 100% случаев синдрома Ангельмана диагностируется выраженная задержка в развитии психики, отсутствие самостоятельной речи или очень скудный словарный запас. Коммуникация осуществляется с помощью мимики, жестов, рисунков. В поведении отмечается гиперактивность, стереотипные движения руками, беспричинный смех.
- Неврологическое обследование. У всех пациентов определяется атаксия и тремор конечностей. 80% больных имеют постнатальную микроцефалию – окружность головы новорожденного меньше 32 см, к 12 месяцам – около 42 см. По данным ЭЭГ выявляется симптоматическая эпилепсия (высокоамплитудные разряды медленных комплексных волн), клинически возможны судорожные припадки. У некоторых детей имеется косоглазие, диффузное снижение мышечного тонуса, усиление сухожильных рефлексов, гиперкинезы.
- Генетическое исследование. Лабораторная диагностика нацелена на выявление мутаций и делеций в гене UBE3A. Последовательно проводится комплекс процедур, включающий флуоресцентную гибридизацию in situ, анализ мутации центра запечатления, метилирование ДНК СА/ПВС региона, диагностику делеции методом микроматричного анализа, поиск мутационных изменений в локусе UBE3A.
Родителям важно внимательно следить за состоянием крохи и при обнаружении первых тревожных сигналов сразу же обращаться за медицинской помощью. Если предварительно диагностирован синдром петрушки заболевание, для подтверждения наличия патологии назначают такое исследование:
Если предварительно диагностирован синдром петрушки заболевание, для подтверждения наличия патологии назначают такое исследование:
- общий осмотр пациента;
- консультация психиатра;
- неврологическое обследование;
- лабораторная диагностика.
Первые признаки синдрома Вильямса
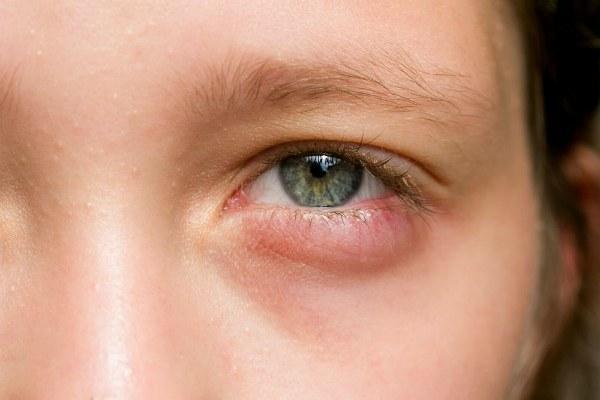
Первыми признаками наличия патологии выступает внешний вид больного. Дети, которые страдают синдромом Вильямса, внешне похожи на эльфа из литературных источников. Потому в простонародье заболевание так и называют — синдром эльфа. У больного присутствует широкий лоб и плоская переносица. Цвет глаз всегда ярко-голубой. Подбородок маленький и несколько заостренный. Люди с патологией обладают широким ртом, что наделяет их улыбкой от уха до уха. Щеки у людей с синдромом эльфа полные и опущенные. Наблюдается расхождение бровей по средней линии.
Внешние признаки переменчивы. Однако их совокупность наглядно демонстрирует проявление мутации. Со временем внешность может измениться. Надбровные дуги способны увеличиться. Иногда наблюдается снижение пастозности лица. Плоская переносица может исчезнуть. Расстояние от верхней губы до кончика носа продолжает сохраняться весьма заметно.
Синдром Вильямса является врожденной патологией. Люди, страдающие заболеванием, нередко рождаются на свет переношенные. Однако ребёнок обладает малым весом. Специфические признаки во внешнем виде заметны уже с первых дней жизни. Ребенок может отказываться от груди. У него наблюдается слабость физиологических рефлексов. Присутствует нарушение процесса глотания и сосания. Наблюдается мышечная слабость.
Дети, страдающие синдромом Вильямса, часто срыгивают. У них нередко возникают запоры и приступы рвоты. Во время первых нескольких лет жизни у ребенка часто диагностируются различные патологии. Впоследствии общее состояние организма нормализуется. На первый план выходит умственная отсталость.
В более старшем возрасте у людей с синдромом Вильямса наблюдаются изменения в голосе. Они обладают низким тембром. Голос немного хриплый. У ребенка с патологией нередко наблюдается косоглазие и нависание верхних век.
У пациентов, у которых выявлена проблема, имеется неправильный прикус. Во время осмотра ротовой полости между зубами наблюдаются широкие промежутки. Присутствует множественный кариес.
Формы синдрома Элерса-Данлоса
Болезнь имеет множество форм:
- Синдром Элерса-Данлоса Тип I (ТИП I ЭДС )
- Синдром Элерса-Данлоса Тип II (ТИП II ЭДС)
- Синдром Элерса-Данлоса III типа (тип ЭДС III и гипермобильности в суставах)
- другие формы (тип IV – тип X)
1 тип синдрома Элерса-Данлоса встречается чаще остальных (40-50%). При таком виде заболевания преобладают симптомы, затрагивающие кожный покров, возможность растяжения которого отходит от нормы в 2-2.5 раз. Данный тип сопровождается наружными кровотечениями и варикозными венами в области нижних конечностей, расшатанностью суставов тела, деформацией скелета. Роды при таком виде заболевания часто происходят преждевременно.
При 2 типе наблюдаются аналогичные признаки, но проявляются они не так сильно. Аномальная гибкость наблюдается только в суставах конечностей, преимущественно стоп и кистей. Растяжимость кожи незначительно отклоняется от нормы, то же и с нарушением работы кровеносных сосудов.
3 тип проявляется ввиду аутосомно-доминантного наследования и протекает доброкачественно. Включает повышенную подвижность суставов по всему телу, деформацию скелета и мышечных тканей и незначительные проявления эластичности кожи.
Редкий и тяжело протекающий 4 тип может быть доминантной и рецессивной. Отличается наличием, у больных повышенной подвижности суставов в пальцах рук, спонтанного возникновения гематом внутренних органов и разрывом всех видов сосудов, что часто приводит к летальным исходам.
5 тип синдрома Элерса-Данлоса происходит из-за X-сцепленного рецессивного наследования. Для него характерны умеренно повышенная мобильность суставов, кровоподтеки и повышенная растяжимость кожи и ее чувствительность.
6 тип наследуется по аутосомно-рецессивному типу. Помимо кровотечений, повышенной подвижности суставов и неестественно эластичной кожи характеризуется увеличением мышечного тонуса (гипертонией мышц), а так же косолапостью и тяжелой формой кифосколиоза, выявляется большой перечень патологий зрения.
7 тип, называемый артроклазией, может наследоваться аутосомно-доминантно и аутосомно-рецессивно. При этом типе у людей часто случаются травмы из-за нездоровой подвижности суставов. Пациентам, подверженным такому типу заболевания, свойственно иметь маленький рост.
8 тип характеризуется аутосомно-доминантным наследованием, и в большей степени для него характерна чувствительность кожи к внешним воздействиям, а так же воспаление тканей, окружающих зубы, что приводит к их ранней потере.
По итогам современных исследований 9 и 10 тип синдрома Элерса-Данлоса не входит в перечень типов классификации заболевания. Для них характерно аутосомно-рецессивное наследование. Кроме гиперэластичности кожи наблюдаются линейные полосы в местах, где кожа наиболее растянута.
Так же присутствует агрегация тромбоцитов и гиперподвижность суставов. У подверженных десятому типу часто наблюдаются врожденные вывихи бедра и постоянно повторяющиеся вывихи плечевых суставов и надколенника.
Факторы-провокаторы синдрома Вильямса
В научных кругах ведутся споры вокруг причины, вызывающей генетическое заболевание. Часть специалистов поддерживает теорию наследственного фактора, остальные склоняются к спонтанной мутации во время зачатия. Механизмом, запускающим патологический процесс, является делеция (утрата) пары в седьмой хромосоме. В состав отсутствующей копии входит больше 25 кодирующих генов, от которых зависит:
- углеводный метаболизм;
- синтез эластина;
- функционирование головного мозга;
- кальциевый обмен;
- состояние нейронов в нервной ткани;
- развитие интеллекта.
Недостаточное производство эластина, входящего в клеточный состав стенок сосудов и сердечного клапана, приводит к появлению пороков органа и гипертензии. Нарушение метаболизма кальция становится причиной гиперкальциемии, при которой истончаются стенки артерий и клапана, вызывая сердечно-сосудистую недостаточность. Дефицит питания нейронов отражается на формировании центральной нервной системы, развитии головного мозга, лицевой дисморфологии.
Вероятность синдрома Вильямса у потомства возрастает, если генетическая мутация диагностировалась у родителей или близких родственников. В этом случае не отмечено выпадение одной копии в наборе на момент зачатия. С учетом клинических проявлений у новорожденного недостающее звено в седьмой хромосоме плод получил путем наследования. Такая причина развития аномалии встречается очень редко.
Более распространенная теория, чем аутосомно-доминантный вариант, утверждает, что синдром Вильямса у детей формируется путем спонтанного нарушения хромосомной цепочки в момент зачатия. Провоцирующим фактором является воздействие на организм одного или обоих родителей:
- вида трудовой деятельности на вредном производстве, связанном с ядовитыми химикатами, тяжелыми металлами, радиацией;
- концентрация токсинов в организме по вине плохой экологии в месте проживания мужчины или женщины;
- употребление алкоголя, наркотиков, курение табака.
В этом случае у родителей существует риск появления на свет ребенка с недостающим звеном в хромосоме.

Борьба с гиперкальциемией
Важно поддерживать должную концентрацию кальция в крови, чтобы избежать опасных осложнений болезни. В период новорождённости и грудном возрасте решается вопрос о необходимости вскармливания малыша специальными смесями с низким содержанием элемента
По мере роста ребёнка рацион его постепенно расширяется, вводятся новые продукты, но контроль за уровнем кальция необходимо осуществлять постоянно.
В целом объём вводимой жидкости, нутриентов должен быть достаточный для обеспечения нормального функционирования организма без перегрузки кальцийсодержащими продуктами. При значительном повышении кальция в крови малыша может потребоваться медикаментозная, инфузионная терапия, введение препаратов калия.
Малышам с синдромом Вильямса противопоказано применение витаминно-минеральных комплексов, в состав которых входит кальций и витамин D.
Лечение сердечно-сосудистой патологии
В зависимости от выраженности проявлений меняется и тактика лечения патологии сердца и сосудов. Незначительные проблемы лечатся консервативно, артериальное давление снижается препаратами из группы ингибиторов АПФ, антагонистов кальция, диуретиков, бета-адреноблокаторов. В тяжёлых случаях требуется хирургическое вмешательство.
Симптоматическая терапия
Дети с синдромом «лица эльфа» состоят на учёте у многих специалистов. Им проводится коррекция остроты зрения, устранение дефектов зубов, лечение плоскостопия и патологий позвоночника, удаление грыж.
Врач-невропалог нередко назначает в комплексном лечении препараты, улучшающие мозговое кровообращение и стимулирующие психическое развитие ребёнка – «Пирацетам», «Винпоцетин», «Церебролизин». Для стимулирования физического развития хорошо использовать методы физиотерапии – массаж, ЛФК, магнито- и ультрозвуковая терапии и другие.
Социальная адаптация ребёнка
Психотерапия, работа с психологами, логопедами необходимы для улучшения интеллектуального развития ребёнка, поддержки семьи. Грамотные специалисты объяснят родителям, какие упражнения способствуют развитию речи, письма, чтения, невербальных навыков. Семьям, в которых рождается необычный ребёнок, требуется помощь психолога, который поможет принять исключительность малыша, научит кроху адаптироваться к окружающим людям.
Особенностью детей с «лицом эльфа» является выраженная разница между вербальным и невербальным уровнем IQ. Интеллектуальное развитие происходит за счёт овладения речью, в то время как пространственные представления не развиваются. Этим детям сложно даются такие задачи, как собрать пирамидку, нарисовать рисунок, освоить математику, выполнить физическое упражнение.
Перспективы лечения синдрома Вильямса

Положительные качества личности у больных с синдромом Вильямса являются одновременно и отрицательными. Высокий уровень гормона окситоцина, вызывающий повышенный интерес к другим людям, а также особенность развития миндалевидного тела головного мозга подавляют способность распознавать отрицательные эмоции и агрессию и правильно интерпретировать проявления приязни и неприязни. Люди с «лицом эльфа» отличаются затруднениями в распознавании отрицательных эмоций даже на фотографиях, что повышает их уровень виктимности, так называемой привлекательности для агрессоров.
Умственная отсталость, распространенность синдрома дефицита внимания, сложности с целостным визуальным восприятием, обработкой информации и логическим мышлением не предполагают полноценного образования. В большинстве случаев специалисты рекомендуют родителям таких детей концентрироваться на обучении бытовым навыкам и, по возможности, выборе рабочих специальностей, не требующих систематических знаний.
Синдром Вильямса не предполагает статуса инвалида или особых льгот. Сопутствующие заболевания, выраженные пороки сердца могут послужить основой для присвоения группы инвалидности, но коррекцией умственного и психического развития «детей-эльфов» занимаются родители, в основном, за счет семейного бюджета. Причем такая ситуация не наблюдается только в России.
Личный опыт
Мария, мама ребенка с синдромом Вильямса, Франция
В обычный детский сад нам разрешено ходить 3 раза в неделю на полтора часа. И два раза в неделю 45-минутные занятия со специалистом, на которые мы ездим в другой город — это все, что положено от государства. Все остальное — логопеды, психологи, бассейны — за наш счет. Дети с синдромом Вильямса инвалидами не признаются.
Я однажды распечатала статью из русскоязычного интернета, где россияне ругают правительство и рассказывают, что положено детям с таким диагнозом во Франции, перевела на французский и дала почитать нашему доктору. Он очень смеялся. Нам положено только бесплатное медицинское обслуживание, коррекции практически нет.
Личный опыт
Мила, США
В Штатах «вильямсятам» инвалидность не положена, только после 18 лет, и то не всем. Так что пока у нас так: есть бесплатная терапия развития до 3 лет, мы начали в год, как получили диагноз, все приходили на дом; бесплатный детский сад по 2,5 ч. в день от 3 лет до 5, бесплатное школьное образование с 5 до 21 года.
Все медицинское обслуживание — по страховке, которую надо покупать, и приходится еще доплачивать в процессе лечения. Пенсию для ребенка выплачивают только нуждающимся семьям, а мы, хотя и живем достаточно скромно по сравнению с местными, не считаемся нуждающимися. Приходится платить.
Препаратов никаких не назначают, считают, что лекарства принимать бесполезно. Даем только витамины.
Медикаментозная коррекция поведения и специфики развития при синдроме Вильямса находится на стадии теоретических исследований. Сегодня в курс терапии рекомендуют включать занятия с логопедом, психологами, интеграционные мероприятия и обучение по программам для детей с нарушениями интеллектуальной деятельности. Для малышей важен контакт со здоровыми детьми, что не всегда достижимо в условиях общеобразовательных школ.
Сказка — ложь, да в ней намек. Музыкальные, дружелюбные и пугливые «эльфы» нуждаются в помощи и поддержке, как медицинской, так и социальной. В России проблемами, связанными с данным диагнозом, занимается благотворительный фонд «Синдром Вильямса» (williams-syndrome.ru), работающий с детьми и их родителями. Взрослые с данным заболеванием, а их, по статистической оценке, около 40000, в основном остаются на попечении родственников или находятся в государственных учреждениях.
Пройдите тестНасколько общителен ребенокС помощью этого теста попробуйте определить уровень коммуникабельности вашего ребенка.
Причины синдрома Вильямса
Причиной синдрома Вильямса является хромосомная аберрация (хромосомная мутация) – делеция участка 7 хромосомы в области ее длинного плеча. Протяженность удаленного участка составляет примерно 1,6-1,8 миллионов пар нуклеотидных оснований, на участке, по разным данным, располагается от 25 до 29 генов. По сути, делеция участка хромосомы равноценна одновременным нонсенс-мутациям множества генов. Наибольшую роль в развитии патологических проявлений при синдроме Вильямса играет отсутствие генов ELN (ген белка эластина), GTF2I (общий фактор транскрипции 2), NCF1 (цитозольный фактор нейтрофилов 1), LIMK1 (кодируемый им белок участвует в формировании цитоскелета нейронов), CGRP (ген кальцитонина). На данном участке 7 хромосомы также расположены такие гены, как GTF1IRD1, GTF2IRD2, GTF2I, BAZ1B, TFII-1 и ряд других – их отсутствие является причиной нарушений углеводного обмена и гиперкальциемии у больных синдромом Вильямса.
В подавляющем большинстве случаев повреждение хромосомы происходит в процессе мейоза при образовании половых клеток, поэтому синдром Вильямса можно рассматривать как результат спонтанной герминативной мутации. Описано несколько случаев аутосомно-доминантного наследования данной патологии. Отсутствие гена ELN в одной из хромосом приводит к недостаточному образованию эластина, главным образом входящего в состав стенок крупных сосудов и клапанов сердца, именно поэтому у больных синдромом Вильямса достаточно часто встречаются пороки сердца и артериальная гипертензия. По некоторым данным, повышенное артериальное давление также может быть вызвано чрезмерной продукцией ангиотензина 2. Неврологические нарушения обусловлены совокупным влиянием отсутствия генов LIMK1, GTF2IRD2 и ряда других, что приводит к патологическим изменениям нейронов и их недостаточному питанию.
Отсутствие гена кальцитонина CGRP становится причиной развития гиперкальциемии, однако, согласно данным современной генетики, у ряда больных синдромом Вильямса обнаруживается его сохранение при повышении уровня кальция в крови. Возможно, гиперкальциемия при этом заболевании обусловлена другими причинами или носит вторичный характер. Совокупность вышеуказанных факторов – пороков сердечно-сосудистой системы, артериальной гипертензии и гиперкальциемии – приводит к значительным нарушениям у больных синдромом Вильямса. К таким нарушениям относят обызвествление стенок сосудов и клапанов сердца, что со временем может стать причиной сердечно-сосудистой недостаточности и смерти больного. Иные нарушения со стороны нервной системы и обмена веществ еще больше усугубляют течение заболевания.
Патологии других органов и систем
 У большинства больных нарушения в работе сердца и сосудов, которые представлены стенозами аорты и артерий различной степени тяжести. При уменьшении просвета почечной артерии половина пациентов страдает высокими показателями артериального давления.
У большинства больных нарушения в работе сердца и сосудов, которые представлены стенозами аорты и артерий различной степени тяжести. При уменьшении просвета почечной артерии половина пациентов страдает высокими показателями артериального давления.
В кровеносном русле наблюдаются значительные количественные показатели кальция, что вызывает психоэмоциональные изменения и болезненное вздутие живота на протяжении первых месяцев жизни.
Пациенты, страдающие заболеванием, имеют низкую массу тела. Набирают они ее намного медленнее, чем их здоровые сверстники. Первый год жизни очень тяжелый по поводу вскармливания. У детей частые позывы на рвоту, нарушены процессы сосания и глотания.
Также могут наблюдаться:
- патологии желудка и кишечного тракта;
- гипофункция щитовидной железы;
- сахарный диабет 2 типа;
- дефекты строения почек;
- грыжи различной локализации;
- низкий тонус мышц.
Чем болеют эльфы?

Большинство младенцев отличаются при рождении низкой массой тела, несоответствием набора веса и роста средним показателям до возраста в 4-5 лет, после чего отмечается скачок роста. Хотя взрослые люди с синдромом Вильямса, как правило, ниже среднего роста.
Для первого года развития характерно повышенное содержание кальция в крови (инфантильная гиперкальциемия). Обычно к 12 месяцам уровень кальция возвращается к норме, хотя может наблюдаться и у взрослых больных.
Из-за недостатка белка эластина у 3 из 4 людей с синдромом Вильямса наблюдаются пороки сердца: обструкция кровотока из правого желудочка в легкие (легочный стеноз), аномальное сужение аорты над сердечным клапаном. Для взрослых характерна гипертония с аномально высокими показателями.
Скелетно-мышечные патологии строения, связанные с синдромом Вильямса, могут включать искривление костей грудины, позвоночного столба (сколиоз или кифоз), врожденный вывих бедра. Также часто встречается высокая подвижность суставов, что в целом определяет характерную для сказок «подпрыгивающую» походку эльфов.
Личный опыт
Марина, мама Миши с синдромом Вильямса
Миша родился очень маленьким, но мы не особенно беспокоились — у мужа от первого брака дочка тоже родилась миниатюрной, а выросла вполне симпатичной девушкой. Когда нам в роддоме показывали, как пеленать и массировать детей, я впервые пригляделась: все младенцы были вполне упитанными, как с картинки, а мой похож на мороженого цыпленка. Но, со слов мужа, дочка была такая же.
Около Мишкиных полугода мы начали разбираться с причиной шумов в сердце, история была долгая, пока мы не попали на прием к профессору. Он посмотрел на результаты обследований и «выдал текст» про наш новый диагноз: и зубы будут кривые, и мелкой моторики можно не ждать, и пойдет Мишка с трудом, и умом будет на уровне детского сада.
Я приходила в себя месяца два после такого прогноза, перечитала в интернете все и постоянно следила за ребенком: за игрушками он следил, мелкие предметы пытался брать, сел и пошел, да, позже, чем положено, но сам. Таблеток никаких мы не принимали, и не назначал никто, ходим к логопеду, в сад в кратковременную группу 3 раза в неделю, а в качестве коррекции все время стараемся поменьше сидеть дома.
Сейчас Мишка физически вполне развит и не оправдал прогнозов профессора. Интеллект, конечно, отстает и в поведении отличается от ровесников. Ну, это уже каждый родитель «вильямсенка» знает.
Что делать?
Если Вы считаете, что у вас Синдром Ангельмана
и характерные для этого заболевания симптомы, то вам могут помочь врачи: генетик, педиатр.
Желаем всем здоровья!
Заболевания со схожими симптомами
Синдром Мартина-Белл (совпадающих симптомов: 5 из 20)
Синдром Мартина-Белл – распространенная генетическая аномалия, которая чаще диагностируется у мальчиков, нежели у девочек. Данные медицинской статистики достаточно неутешительны – частота рождения малышей с таким недугом составляет 1 на 6000 девочек, и 1 на 4000 мальчиков.
Стоит отметить, что у представителей сильного пола заболевание протекает более тяжело, но все же бывают и некоторые исключения. Причиной проявления у девочек более стертой симптоматики является компенсирующий эффект, которые оказывает вторая Х-хромосома в кариотипе.
Приобретенный талант
Одно или даже несколько из перечисленных качеств, по всей видимости, может получить любой человек без больших усилий со своей стороны, если ему «повезет». Как получить синдром саванта? Достаточно, например, получить тупым предметом в переулке по голове, неудачно нырнуть в бассейн, перенести геморрагический инсульт или, конечно же, попасть под удар молнии. Однако, вероятность такого «везения» не высчитает и савант, так как голову ежедневно травмируют тысячи, а способностями к математике и рисованию после этого блеснул пока только продавец матрацев Джейсон Пэджетт, сильно избитый сентябрьской ночью 2002 г. на выходе из караоке-бара. Помимо проблем со здоровьем, бывший 31‑летний любитель выпить и попеть приобрел «геометрическое мышление». Он стал видеть окружающий мир в целом и математические формулы в частности как совокупность линий и геометрических фигур. В итоге Пэджетт профессионально занялся теорией чисел.
Описан случай 9‑летнего мексиканского мальчика, раненного пулей в левую височную область при ограблении. Помимо того, что у него случился правосторонний паралич, он еще разучился говорить, читать, писать и считать. В плюсе: перестал теряться в незнакомой местности (всегда знал дорогу к новому дому при частых переездах семьи), начал отлично чинить и совершенствовать всевозможные механические устройства и с фотографической точностью перерисовывать увиденное.
Возрастные дегенеративные заболевания головного мозга обычно ставят крест на карьере художника. Однако бывает и наоборот: в 1998 г. Брюс Миллер с соавторами описали в журнале Neurology пять случаев внезапного желания и умения рисовать у пациентов с лобно-височной деменцией, причём навык развивался параллельно с тяжестью заболевания. При проведении однофотонной эмиссионной компьютерной томографии у пациентов было выявлено преимущественное поражение левых отделов мозга.
Симптомы и признаки
Клиническая картина недуга весьма разнообразна и может отличаться в каждом конкретном случае. В частности, у ребенка проявляются физические и психические отклонения, некоторые признаки характерны для всех случаев, другие встречаются значительно реже.
| Физические отклонения | Психические отклонения |
|
|
Клинические проявления недуга принято делить также в зависимости от их распространенности. Так, существуют симптомы, которые встречаются у всех детей, страдающих данным недугом, признаки, возникающие в большинстве случаев (но не всегда) и более редкие проявления.
| Характерные симптомы | Частые признаки | Более редкие проявления |
|
|
|
Клинические проявления недуга меняются по мере взросления ребенка. Так, у детей подросткового возраста наблюдается задержка полового созревания (при этом детородная функция в дальнейшем не нарушается), а когда ребенок становится взрослым, он выглядит значительно моложе своих сверстников.
Нередко у детей старшего возраста возникают и другие проявления заболевания, такие как нарушение мочеиспускания, сложные нарушения моторики рук, проблемы с общением (подросток понимает устную речь, но не видит необходимости отвечать собеседнику).